Rebetol 200mg Ribavirin Uso, efeitos colaterais e dosagem. Preço na farmácia online. Medicamentos genericos sem receita.
O que é Rebetol 200mg e como é usado?
O Rebetol é um medicamento de prescrição utilizado para tratar os sintomas da Hepatite C Crónica. O Rebetol 200mg pode ser utilizado sozinho ou com outros medicamentos.
O Rebetol pertence a uma classe de medicamentos denominados Agentes da Hepatite B/Hepatite C; Agentes RSV.
Não se sabe se Rebetol 200mg é seguro e eficaz em crianças com menos de 3 anos de idade.
Quais são os possíveis efeitos colaterais do Rebetol?
Rebetol pode causar efeitos colaterais graves, incluindo:
- urticária,
- dificuldade para respirar,
- inchaço do rosto, lábios, língua ou garganta,
- problemas de visão
- dor intensa na parte superior do estômago se espalhando para as costas,
- náusea,
- vômito,
- diarréia,
- tosse nova ou agravada,
- febre,
- dor no peito lancinante,
- chiado,
- falta de ar,
- Depressão severa,
- pensamentos de automutilação,
- pensamentos de ferir os outros,
- pele pálida ou amarelada,
- urina de cor escura,
- confusão,
- fraqueza,
- arrepios,
- sintomas como os da gripe,
- gengivas inflamadas,
- aftas,
- feridas na pele,
- contusões fáceis,
- sangramento incomum e
- tontura
- Obtenha ajuda médica imediatamente, se tiver algum dos sintomas listados acima.
- Os efeitos colaterais mais comuns do Rebetol incluem:
- náusea,
- vômito,
- perda de apetite,
- febre,
- arrepios,
- tremendo,
- baixa contagem de células sanguíneas,
- anemia,
- fraqueza,
- cansaço,
- dor de cabeça,
- dor muscular,
- mudanca de humor,
- ansiedade, e
- irritabilidade
Informe o médico se tiver algum efeito colateral que o incomode ou que não desapareça.
Estes não são todos os efeitos secundários possíveis de Rebetol. Para mais informações, consulte seu médico ou farmacêutico.
Ligue para o seu médico para aconselhamento médico sobre os efeitos colaterais. Você pode relatar efeitos colaterais ao FDA em 1-800-FDA-1088.
AVISO
RISCO DE TRANSTORNOS GRAVES E EFEITOS ASSOCIADOS À RIBAVIRINA
- REBETOL em monoterapia não é eficaz no tratamento da infecção crônica pelo vírus da hepatite C e não deve ser usado isoladamente para esta indicação [ver ADVERTÊNCIAS E PRECAUÇÕES].
- A toxicidade primária da ribavirina é a anemia hemolítica. A anemia associada à terapia com REBETOL 200mg pode resultar no agravamento da doença cardíaca que levou a infartos do miocárdio fatais e não fatais. Pacientes com história de doença cardíaca significativa ou instável não devem ser tratados com REBETOL [ver POSOLOGIA E ADMINISTRAÇÃO, ADVERTÊNCIAS E PRECAUÇÕES e REAÇÕES ADVERSAS].
- Efeitos teratogênicos e embriocidas significativos foram demonstrados em todas as espécies animais expostas à ribavirina. Além disso, a ribavirina tem meia-vida de dose múltipla de 12 dias e, portanto, pode persistir em compartimentos não plasmáticos por até 6 meses. Portanto, a terapia com REBETOL é contraindicada em mulheres grávidas e nos parceiros masculinos de mulheres grávidas. Deve-se ter extremo cuidado para evitar a gravidez durante a terapia e por 6 meses após a conclusão do tratamento em pacientes do sexo feminino e em parceiras de pacientes do sexo masculino que estejam recebendo terapia com REBETOL. Pelo menos duas formas confiáveis de contracepção eficaz devem ser utilizadas durante o tratamento e durante o período de acompanhamento de 6 meses após o tratamento [ver CONTRA-INDICAÇÕES, ADVERTÊNCIAS E PRECAUÇÕES, Uso em populações específicas, Toxicologia não clínica e INFORMAÇÕES AO PACIENTE].
DESCRIÇÃO
REBETOL (ribavirina), é um análogo de nucleosídeo sintético (análogo de purina). O nome químico da ribavirina é 1-β-D-ribofuranosil-1H-1,2,4-triazol-3-carboxamida e tem a seguinte fórmula estrutural (ver Figura 1).
Figura 1: Fórmula Estrutural
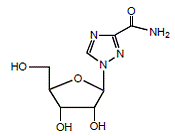
A ribavirina é um pó branco e cristalino. É livremente solúvel em água e ligeiramente solúvel em álcool anidro. A fórmula empírica é C8H12N4O5 e o peso molecular é 244,21.
As cápsulas de REBETOL consistem em um pó branco em uma cápsula de gelatina branca e opaca. Cada cápsula contém 200 mg de ribavirina e os ingredientes inativos celulose microcristalina, lactose mono-hidratada, croscarmelose sódica e estearato de magnésio. O invólucro da cápsula consiste em gelatina, lauril sulfato de sódio, dióxido de silício e dióxido de titânio. A cápsula é impressa com tinta farmacêutica azul comestível que é feita de goma-laca, álcool etílico anidro, álcool isopropílico, álcool n-butílico, propilenoglicol, hidróxido de amônio e laca de alumínio FD&C Blue #2.
REBETOL 200mg solução oral é um líquido límpido, incolor a pálido ou amarelo claro com sabor de chiclete. Cada mililitro da solução contém 40 mg de ribavirina e os ingredientes inativos sacarose, glicerina, sorbitol, propilenoglicol, citrato de sódio, ácido cítrico, benzoato de sódio, aroma natural e artificial para chiclete #15864 e água.
INDICAÇÕES
Hepatite C Crônica (CHC)
REBETOL® (ribavirina) em combinação com interferon alfa-2b (peguilado e não peguilado) é indicado para o tratamento da hepatite C crônica (HCC) em pacientes com 3 anos de idade ou mais com doença hepática compensada [ver AVISOS E PRECAUÇÕES , e Uso em populações específicas ].
Os seguintes pontos devem ser considerados ao iniciar a terapia combinada de REBETOL com PegIntron® ou INTRON A®:
- Essas indicações são baseadas na obtenção de HCV-RNA indetectável após o tratamento por 24 ou 48 semanas e na manutenção de uma Resposta Virológica Sustentada (SVR) 24 semanas após a última dose.
- A terapia combinada com REBETOL/PegIntron é preferível a REBETOL/INTRON A, pois essa combinação fornece taxas de resposta substancialmente melhores [ver Estudos clínicos ].
- Pacientes com as seguintes características são menos propensos a se beneficiar do retratamento após falha de um curso de terapia: não resposta anterior, tratamento anterior com interferon peguilado, fibrose em ponte significativa ou cirrose e infecção pelo genótipo 1 [ver Estudos clínicos ].
- Não estão disponíveis dados de segurança e eficácia para tratamentos superiores a um ano.
DOSAGEM E ADMINISTRAÇÃO
Sob nenhuma circunstância as cápsulas de REBETOL devem ser abertas, esmagadas ou quebradas. REBETOL 200mg deve ser tomado com alimentos [ver FARMACOLOGIA CLÍNICA ]. REBETOL não deve ser usado em pacientes com clearance de creatinina menor que 50 mL/min.
Terapia combinada REBETOL/PegIntron
Pacientes adultos
dose recomendada de PegIntron é de 1,5 mcg/kg/semana por via subcutânea em combinação com 800 a 1400 mg de REBETOL 200 mg cápsulas por via oral com base no peso corporal do paciente (ver Tabela 1). O volume de PegIntron a ser injetado depende da dosagem de PegIntron e do peso corporal do paciente; consulte a bula do PegIntron para obter informações adicionais sobre a dosagem.
Duração do Tratamento - Pacientes virgens de Interferon Alfa
A duração do tratamento para pacientes com genótipo 1 é de 48 semanas. A descontinuação da terapia deve ser considerada em pacientes que não atingem pelo menos uma queda de 2 log10 ou perda de HCV-RNA em 12 semanas, ou se HCV-RNA permanecer detectável após 24 semanas de terapia. Pacientes com genótipo 2 e 3 devem ser tratados por 24 semanas.
Duração do Tratamento - Retratamento com PegIntron/REBETOL de Falhas de Tratamento Anterior
duração do tratamento para pacientes que falharam anteriormente é de 48 semanas, independentemente do genótipo do HCV. Pacientes tratados novamente que não atingem o HCV-RNA indetectável na Semana 12 de terapia, ou cujo HCV-RNA permanece detectável após 24 semanas de terapia, são altamente improváveis de atingir RVS e a descontinuação da terapia deve ser considerada [ver Estudos clínicos ].
Pacientes pediátricos
A dosagem para pacientes pediátricos é determinada pela área de superfície corporal para PegIntron e pelo peso corporal para REBETOL. A dose recomendada de PegIntron é de 60 mcg/m²/semana por via subcutânea em combinação com 15 mg/kg/dia de REBETOL 200 mg por via oral em duas doses divididas (ver Tabela 2) para pacientes pediátricos com idades entre 3-17 anos. Os pacientes que completam 18 anos enquanto recebem PegIntron/REBETOL 200mg devem permanecer no regime posológico pediátrico. A duração do tratamento para pacientes com genótipo 1 é de 48 semanas. Pacientes com genótipo 2 e 3 devem ser tratados por 24 semanas.
Terapia combinada REBETOL/INTRON A
Adultos
Duração do Tratamento - Pacientes virgens de Interferon Alfa
dose recomendada de INTRON A é de 3 milhões de UI três vezes por semana por via subcutânea. A dose recomendada de REBETOL cápsulas depende do peso corporal do paciente (consulte a Tabela 3). A duração recomendada do tratamento para pacientes não tratados previamente com interferon é de 24 a 48 semanas. A duração do tratamento deve ser individualizada para o paciente, dependendo das características basais da doença, resposta à terapia e tolerabilidade do regime [ver INDICAÇÕES E USO , REAÇÕES ADVERSAS , e Estudos clínicos ]. Após 24 semanas de tratamento, a resposta virológica deve ser avaliada. A descontinuação do tratamento deve ser considerada em qualquer paciente que não tenha atingido um HCV-RNA abaixo do limite de detecção do ensaio em 24 semanas. Não existem dados de segurança e eficácia do tratamento por mais de 48 semanas na população de pacientes não tratados anteriormente.
Duração do Tratamento - Retratamento com INTRON A/REBETOL em Pacientes com Recaída
Em pacientes que recidivam após monoterapia com interferon não peguilado, a duração recomendada do tratamento é de 24 semanas.
Pediatria
A dose recomendada de REBETOL é de 15 mg/kg por dia por via oral (dose dividida AM e PM). Consulte a Tabela 2 para a dosagem pediátrica de REBETOL em combinação com INTRON A. INTRON A para injeção por peso corporal de 25 kg a 61 kg é de 3 milhões de UI/m² três vezes por semana por via subcutânea. Consulte a tabela de dosagem para adultos para peso corporal superior a 61 kg.
A duração recomendada do tratamento é de 48 semanas para pacientes pediátricos com genótipo 1. Após 24 semanas de tratamento, a resposta virológica deve ser avaliada. A descontinuação do tratamento deve ser considerada em qualquer paciente que não tenha atingido um HCV-RNA abaixo do limite de detecção do ensaio até este momento. A duração recomendada do tratamento para pacientes pediátricos com genótipo 2/3 é de 24 semanas.
Testes laboratoriais
Os seguintes exames laboratoriais são recomendados para todos os pacientes tratados com REBETOL 200mg, antes do início do tratamento e depois periodicamente.
Testes hematológicos padrão - incluindo hemoglobina (pré-tratamento, Semana 2 e Semana 4 de terapia, e conforme clinicamente apropriado [ver AVISOS E PRECAUÇÕES ], contagens completas e diferenciais de leucócitos e contagem de plaquetas.
- Química do sangue - testes de função hepática e TSH.
- Gravidez - incluindo acompanhamento mensal para mulheres em idade fértil.
- ECG [ver AVISOS E PRECAUÇÕES ].
Modificações de dose
Se reações adversas graves ou anormalidades laboratoriais se desenvolverem durante a terapia combinada com REBETOL/INTRON A ou terapia com REBETOL/PegIntron, modifique ou descontinue a dose até que a reação adversa diminua ou diminua em gravidade [ver AVISOS E PRECAUÇÕES ]. Se a intolerância persistir após o ajuste da dose, a terapia combinada deve ser descontinuada. A redução da dose de PegIntron em pacientes adultos em terapia combinada de REBETOL/PegIntron é realizada em um processo de duas etapas, desde a dose inicial original de 1,5 mcg/kg/semana, para 1 mcg/kg/semana, depois para 0,5 mcg/kg/semana , se necessário. Consulte a bula do PegIntron para obter informações adicionais sobre a redução da dose de PegIntron.
No estudo 2 de terapia combinada para adultos, ocorreram reduções de dose em 42% dos indivíduos que receberam PegIntron 1,5 mcg/kg e REBETOL 800 mg diariamente, incluindo 57% dos indivíduos com peso igual ou inferior a 60 kg. No Estudo 4, 16% dos indivíduos tiveram uma redução da dose de PegIntron para 1 mcg/kg em combinação com REBETOL, com um adicional de 4% exigindo a segunda redução da dose de PegIntron para 0,5 mcg/kg devido a eventos adversos [ver REAÇÕES ADVERSAS ].
redução da dose em pacientes pediátricos é conseguida modificando a dose recomendada de PegIntron em um processo de duas etapas da dose inicial original de 60 mcg/m²/semana, para 40 mcg/m²/semana, depois para 20 mcg/m²/semana, se necessário (ver Tabela 4). No ensaio de terapia combinada pediátrica, ocorreram reduções de dose em 25% dos indivíduos que receberam PegIntron 60 mcg/m² semanalmente e REBETOL 15 mg/kg diariamente. A redução da dose em pacientes pediátricos é conseguida modificando a dose recomendada de REBETOL da dose inicial inicial de 15 mg/kg/dia em um processo de duas etapas para 12 mg/kg/dia, depois para 8 mg/kg/dia, se necessário ( ver Tabela 4).
REBETOL 200mg não deve ser usado em pacientes com clearance de creatinina menor que 50 mL/min. Pacientes com função renal comprometida e aqueles com mais de 50 anos devem ser cuidadosamente monitorados em relação ao desenvolvimento de anemia [ver AVISOS E PRECAUÇÕES , Uso em populações específicas , e FARMACOLOGIA CLÍNICA ].
REBETOL 200mg deve ser administrado com cautela a pacientes com doença cardíaca pré-existente. Os pacientes devem ser avaliados antes do início da terapia e devem ser monitorados adequadamente durante a terapia. Se houver qualquer deterioração do estado cardiovascular, a terapia deve ser interrompida [ver AVISOS E PRECAUÇÕES ].
Para pacientes com histórico de doença cardiovascular estável, é necessária uma redução permanente da dose se a hemoglobina diminuir em mais ou igual a 2 g/dL durante qualquer período de 4 semanas. Além disso, para esses pacientes com história cardíaca, se a hemoglobina permanecer inferior a 12 g/dL após 4 semanas com uma dose reduzida, o paciente deve descontinuar a terapia combinada.
Recomenda-se que um paciente cujo nível de hemoglobina caia abaixo de 10 g/dL tenha sua dose de REBETOL modificada ou descontinuada de acordo com a Tabela 4 [ver AVISOS E PRECAUÇÕES ].
Consulte a bula de INTRON A ou PegIntron para obter informações adicionais sobre como reduzir uma dose de INTRON A ou PegIntron.
Descontinuação da dosagem
Adultos
No genótipo 1 do HCV, pacientes virgens de interferon alfa recebendo PegIntron em combinação com ribavirina, a descontinuação da terapia é recomendada se não houver uma queda de pelo menos 2 log10 ou perda de HCV-RNA em 12 semanas de terapia, ou se HCV-RNA os níveis permanecem detectáveis após 24 semanas de terapia. Independentemente do genótipo, é altamente improvável que pacientes tratados anteriormente que tenham RNA-VHC detectável na Semana 12 ou 24 atinjam RVS e a descontinuação da terapia deve ser considerada.
Pediatria (3-17 anos de idade)
Recomenda-se que os doentes a receber a combinação PegIntron/REBETOL (excluindo HCV Genótipo 2 e 3) sejam descontinuados da terapêutica às 12 semanas se o seu tratamento Semana 12 HCV-RNA descer menos de 2 log10 em comparação com um pré-tratamento ou às 24 semanas se tiverem sintomas detectáveis HCV-RNA na semana de tratamento 24.
COMO FORNECIDO
Formas de dosagem e pontos fortes
REBETOL 200mg cápsulas 200mg
REBETOL 200mg Solução Oral 40mg por mL
Armazenamento e manuseio
REBETOL 200 mg cápsulas são cápsulas brancas e opacas com REBETOL, 200 mg, e o logotipo da Schering Corporation impresso no invólucro da cápsula; as cápsulas são acondicionadas em um frasco contendo 56 cápsulas ( NDC 0085-1351-05), 70 cápsulas ( NDC 0085-1385-07), e 84 cápsulas ( NDC 0085-1194-03).
REBETOL 200mg Solução Oral 40mg por mL é um líquido com sabor de chiclete transparente, incolor a pálido ou amarelo claro e é embalado em frascos de vidro âmbar de 4 onças (100 mL/frasco) com tampas à prova de crianças ( NDC 0085-1318-01).
O frasco de REBETOL Cápsulas deve ser armazenado a 25°C (77°F); excursões permitidas para 15-30°C (59-86°F) [ver Temperatura ambiente controlada USP ].
REBETOL 200mg Solução Oral deve ser armazenado entre 2-8°C (36-46°F) ou a 25°C (77°F); excursões permitidas para 15-30°C (59-86°F) [ver Temperatura ambiente controlada USP ].
REBETOL 200mg Solução Oral fabricada para: Merck Sharp & Dohme Corp., uma subsidiária da Whitehouse Station, NJ 08889, EUA. Fabricado por: Schering-Plough Canada, Inc., Pointe Claire, Quebec, Canadá Cápsulas REBETOL fabricadas por: Merck Sharp & Dohme Corp., uma subsidiária da Whitehouse Station, NJ 08889, EUA. Revisado: dezembro de 2014.
EFEITOS COLATERAIS
Ensaios clínicos com REBETOL em combinação com PegIntron ou INTRON A foram conduzidos em mais de 7.800 indivíduos de 3 a 76 anos de idade.
A toxicidade primária da ribavirina é a anemia hemolítica. Reduções nos níveis de hemoglobina ocorreram nas primeiras 1 a 2 semanas de terapia oral. Reações cardíacas e pulmonares associadas à anemia ocorreram em aproximadamente 10% dos pacientes [ver AVISOS E PRECAUÇÕES ].
Mais de 96% de todos os indivíduos em ensaios clínicos experimentaram uma ou mais reações adversas. As reações adversas mais comumente relatadas em indivíduos adultos recebendo PegIntron ou INTRON A em combinação com REBETOL 200mg foram inflamação/reação no local da injeção, fadiga/astenia, dor de cabeça, calafrios, febre, náusea, mialgia e ansiedade/labilidade emocional/irritabilidade. As reações adversas mais comuns em pacientes pediátricos, com 3 anos ou mais, recebendo REBETOL 200 mg em combinação com PegIntron ou INTRON A foram pirexia, cefaleia, neutropenia, fadiga, anorexia, eritema no local da injeção e vômitos.
A seção Reações Adversas faz referência aos seguintes ensaios clínicos:
- Ensaios de terapia combinada REBETOL/PegIntron:
- Estudo Clínico 1 – avaliou a monoterapia com PegIntron (não descrita neste rótulo; consulte a rotulagem do PegIntron para obter informações sobre este estudo).
- Estudo 2 – avaliou a dose fixa de 800 mg/dia de REBETOL em combinação com PegIntron 1,5 mcg/kg/semana ou com INTRON A.
- Estudo 3 – avaliou PegIntron/rebetol baseado no peso 200mg em combinação com regime de PegIntron/dose plana REBETOL 200mg.
- Estudo 4 – comparou duas doses de PegIntron (1,5 mcg/kg/semana e 1 mcg/kg/semana) em combinação com REBETOL e um terceiro grupo de tratamento recebendo Pegasys® (180 mcg/semana)/Copegus® (1000-1200 mg/dia ).
- Estudo 5 – avaliou PegIntron (1,5 mcg/kg/semana) em combinação com REBETOL baseado no peso em indivíduos com falha de tratamento anterior.
- Terapia combinada PegIntron/REBETOL 200mg em pacientes pediátricos
- Ensaios de Terapia Combinada REBETOL/INTRON A para adultos e pediatria
Reações adversas graves ocorreram em aproximadamente 12% dos indivíduos em ensaios clínicos com PegIntron com ou sem REBETOL [ver AVISO EM CAIXA , AVISOS E PRECAUÇÕES ]. Os eventos graves mais comuns ocorridos em indivíduos tratados com PegIntron e REBETOL 200mg foram depressão e ideação suicida [ver AVISOS E PRECAUÇÕES ], cada um ocorrendo com uma frequência inferior a 1%. Ideação ou tentativas suicidas ocorreram com mais frequência entre pacientes pediátricos, principalmente adolescentes, em comparação com pacientes adultos (2,4% versus 1%) durante o tratamento e acompanhamento fora da terapia [ver AVISOS E PRECAUÇÕES ]. A reação fatal mais comum que ocorreu em indivíduos tratados com PegIntron e REBETOL 200 mg foi parada cardíaca, ideação suicida e tentativa de suicídio [ver AVISOS E PRECAUÇÕES ], todos ocorrendo em menos de 1% dos indivíduos.
Como os ensaios clínicos são conduzidos em condições muito variadas, as taxas de reações adversas observadas nos ensaios clínicos de um medicamento não podem ser diretamente comparadas às taxas nos ensaios clínicos de outro medicamento e podem não refletir as taxas observadas na prática clínica.
Experiência em Ensaios Clínicos - Terapia Combinada REBETOL/PegIntron
Assuntos adultos
As reações adversas que ocorreram no ensaio clínico com incidência superior a 5% são fornecidas pelo grupo de tratamento da Terapia de Combinação REBETOL/PegIntron (Estudo 2) na Tabela 5.
A Tabela 6 resume as reações adversas relacionadas ao tratamento no Estudo 4 que ocorreram com uma incidência maior ou igual a 10%.
A incidência de reações adversas graves foi comparável em todos os ensaios. No Estudo 3, houve uma incidência semelhante de reações adversas graves relatadas para o grupo REBETOL baseado no peso (12%) e para o regime de dose plana de REBETOL. No Estudo 2, a incidência de reações adversas graves foi de 17% nos grupos PegIntron/REBETOL em comparação com 14% no grupo INTRON A/REBETOL.
Em muitos, mas não em todos os casos, as reações adversas foram resolvidas após a redução da dose ou descontinuação da terapia. Alguns indivíduos experimentaram reações adversas graves contínuas ou novas durante o período de acompanhamento de 6 meses. No Estudo 2, muitos indivíduos continuaram a apresentar reações adversas vários meses após a descontinuação da terapia. Ao final do período de acompanhamento de 6 meses, a incidência de reações adversas contínuas por classe corporal no grupo PegIntron 1.5/REBETOL 200mg foi de 33% (psiquiátrico), 20% (musculoesquelético) e 10% (para endócrino e para GI). Em aproximadamente 10 a 15% dos indivíduos, a perda de peso, fadiga e dor de cabeça não foram resolvidas.
Houve 31 mortes de indivíduos que ocorreram durante o tratamento ou durante o acompanhamento nestes ensaios clínicos. No Estudo 1, houve 1 suicídio em um indivíduo que recebeu a monoterapia com PegIntron e 2 mortes entre os indivíduos que receberam a monoterapia com INTRON A (1 assassinato/suicídio e 1 morte súbita). No Estudo 2, houve 1 suicídio em um indivíduo recebendo terapia combinada PegIntron/REBETOL 200mg; e 1 óbito no grupo INTRON A/REBETOL 200mg (acidente automobilístico). No Estudo 3, houve 14 mortes, 2 das quais foram prováveis suicídios e 1 foi uma morte inexplicável em uma pessoa com histórico médico relevante de depressão. No Estudo 4, houve 12 mortes, 6 das quais ocorreram em indivíduos que receberam a terapia combinada PegIntron/REBETOL, 5 no braço PegIntron 1,5 mcg/REBETOL (N=1019) e 1 no braço PegIntron 1 mcg/REBETOL (N= 1016), e 6 dos quais ocorreram em indivíduos que receberam Pegasys/Copegus (N=1035); houve 3 suicídios que ocorreram durante o período de acompanhamento sem tratamento em indivíduos que receberam a terapia combinada PegIntron (1,5 mcg/kg)/REBETOL 200mg.
Nos estudos 1 e 2, 10 a 14% dos indivíduos que receberam PegIntron, isoladamente ou em combinação com REBETOL 200mg, interromperam a terapia em comparação com 6% tratados com INTRON A isolado e 13% tratados com INTRON A em combinação com REBETOL. Da mesma forma, no Estudo 3, 15% dos indivíduos que receberam PegIntron em combinação com REBETOL baseado no peso e 14% dos indivíduos que receberam PegIntron e REBETOL em dose plana descontinuaram a terapia devido a uma reação adversa. As razões mais comuns para a descontinuação da terapia foram relacionadas a efeitos conhecidos do interferon de reações adversas psiquiátricas, sistêmicas (por exemplo, fadiga, dor de cabeça) ou gastrointestinais. No Estudo 4, 13% dos indivíduos no braço PegIntron 1,5 mcg/REBETOL 200mg, 10% no braço PegIntron 1 mcg/REBETOL 200mg e 13% no braço Pegasys 180 mcg/Copegus descontinuaram devido a eventos adversos.
No Estudo 2, as reduções de dose devido a reações adversas ocorreram em 42% dos indivíduos que receberam PegIntron (1,5 mcg/kg)/REBETOL 200mg e em 34% daqueles que receberam INTRON A/REBETOL. A maioria dos indivíduos (57%) com peso igual ou inferior a 60 kg a receber PegIntron (1,5 mcg/kg)/REBETOL necessitou de redução da dose. A redução de interferon foi relacionada à dose (PegIntron 1,5 mcg/kg maior que PegIntron 0,5 mcg/kg ou INTRON A), 40%, 27%, 28%, respectivamente. A redução da dose de REBETOL 200mg foi semelhante nos três grupos, 33 a 35%. As razões mais comuns para modificações de dose foram neutropenia (18%) ou anemia (9%) (ver Valores de laboratório ). Outras razões comuns incluem depressão, fadiga, náusea e trombocitopenia. No Estudo 3, as modificações de dose devido a reações adversas ocorreram com mais frequência com a dosagem baseada no peso (WBD) em comparação com a dosagem plana (29% e 23%, respectivamente). No Estudo 4, 16% dos indivíduos tiveram uma redução da dose de PegIntron para 1 mcg/kg em combinação com REBETOL 200mg, com um adicional de 4% exigindo a segunda redução da dose de PegIntron para 0,5 mcg/kg devido a eventos adversos em comparação com 15% de indivíduos no braço Pegasys/Copegus, que necessitaram de uma redução da dose para 135 mcg/semana com Pegasys, com um adicional de 7% no braço Pegasys/Copegus necessitando de uma segunda redução da dose para 90 mcg/semana com Pegasys.
Nos ensaios da combinação PegIntron/REBETOL 200mg, as reações adversas mais comuns foram psiquiátricas, que ocorreram em 77% dos indivíduos no Estudo 2 e 68% a 69% dos indivíduos no Estudo 3. Essas reações adversas psiquiátricas incluíram mais comumente depressão, irritabilidade e insônia, cada uma relatada por aproximadamente 30% a 40% dos indivíduos em todos os grupos de tratamento. Comportamento suicida (ideação, tentativas e suicídios) ocorreu em 2% de todos os indivíduos durante o tratamento ou durante o acompanhamento após a interrupção do tratamento [ver AVISOS E PRECAUÇÕES ]. No Estudo 4, as reações adversas psiquiátricas ocorreram em 58% dos indivíduos no braço PegIntron 1,5 mcg/REBETOL 200mg, 55% dos indivíduos no braço PegIntron 1 mcg/REBETOL 200mg e 57% dos indivíduos no braço Pegasys 180 mcg/Copegus .
PegIntron induziu fadiga ou dor de cabeça em aproximadamente dois terços dos indivíduos, com febre ou calafrios em aproximadamente metade dos indivíduos. A gravidade de alguns desses sintomas sistêmicos (por exemplo, febre e dor de cabeça) tendeu a diminuir com a continuação do tratamento. Nos estudos 1 e 2, inflamação e reação no local de aplicação (por exemplo, hematoma, coceira e irritação) ocorreram em aproximadamente o dobro da incidência com terapias com PegIntron (em até 75% dos indivíduos) em comparação com INTRON A. No entanto, a dor no local de injeção foi pouco frequente (2 a 3%) em todos os grupos. No Estudo 3, houve uma incidência geral de 23% a 24% para reações ou inflamação no local da injeção.
Indivíduos que receberam REBETOL/PegIntron como retratamento após falha em um regime anterior de combinação de interferon relataram reações adversas semelhantes àquelas previamente associadas a este regime durante ensaios clínicos de indivíduos virgens de tratamento.
Sujeitos Pediátricos
Em geral, o perfil de reações adversas na população pediátrica foi semelhante ao observado em adultos. No ensaio pediátrico, as reações adversas mais prevalentes em todos os indivíduos foram pirexia (80%), dor de cabeça (62%), neutropenia (33%), fadiga (30%), anorexia (29%), eritema no local da injeção (29 %) e vômitos (27%). A maioria das reações adversas relatadas no estudo foi de gravidade leve ou moderada. Reações adversas graves foram relatadas em 7% (8/107) de todos os indivíduos e incluíram dor no local da injeção (1%), dor nas extremidades (1%), dor de cabeça (1%), neutropenia (1%) e pirexia (4). %). As reações adversas importantes que ocorreram nesta população de indivíduos foram nervosismo (7%; 7/107), agressão (3%; 3/107), raiva (2%; 2/107) e depressão (1%; 1/107) . Cinco indivíduos receberam tratamento com levotiroxina, três com hipotireoidismo clínico e dois com elevações de TSH assintomáticas. O ganho de peso e altura de indivíduos pediátricos tratados com PegIntron mais REBETOL ficou abaixo do previsto pelos dados normativos da população para toda a duração do tratamento. Velocidade de crescimento severamente inibida (menos de 3º percentil) foi observada em 70% dos indivíduos durante o tratamento.
Modificações de dose de PegIntron e/ou ribavirina foram necessárias em 25% dos indivíduos devido a reações adversas relacionadas ao tratamento, mais comumente para anemia, neutropenia e perda de peso. Dois indivíduos (2%; 2/107) interromperam a terapia como resultado de uma reação adversa.
As reações adversas que ocorreram com uma incidência maior ou igual a 10% nos indivíduos do estudo pediátrico são fornecidas na Tabela 7.
Noventa e quatro dos 107 indivíduos inscritos em um estudo de acompanhamento de longo prazo de 5 anos. Os efeitos a longo prazo no crescimento foram menores nos indivíduos tratados por 24 semanas do que naqueles tratados por 48 semanas. Vinte e quatro por cento dos indivíduos (11/46) tratados por 24 semanas e 40% dos indivíduos (19/48) tratados por 48 semanas tiveram uma diminuição > 15 percentil de altura para idade desde o pré-tratamento até o final de 5 anos acompanhamento de longo prazo em comparação com os percentis basais pré-tratamento. Onze por cento dos indivíduos (5/46) tratados por 24 semanas e 13% dos indivíduos (6/48) tratados por 48 semanas apresentaram uma diminuição da linha de base pré-tratamento de > 30 percentis de altura para idade até o final do seguimento de 5 anos a longo prazo. Embora observado em todas as faixas etárias, o maior risco de redução da altura no final do acompanhamento de longo prazo pareceu correlacionar-se com o início da terapia combinada durante os anos de pico de velocidade de crescimento esperado. [Ver AVISOS E PRECAUÇÕES ]
Valores de laboratório
Sujeitos Adultos e Pediátricos
perfil de reações adversas no Estudo 3, que comparou a combinação PegIntron/REBETOL 200mg baseado no peso a um regime PegIntron/dose plana REBETOL 200mg, revelou uma taxa aumentada de anemia com a dosagem baseada no peso (29% vs. 19% para o regime baseado no peso vs. regimes de dose plana, respectivamente). No entanto, a maioria dos casos de anemia foi leve e respondeu às reduções de dose.
As alterações nos valores laboratoriais selecionados durante o tratamento em combinação com o tratamento com REBETOL 200mg são descritas abaixo. A diminuição da hemoglobina, leucócitos, neutrófilos e plaquetas pode exigir redução da dose ou descontinuação permanente da terapia [Vejo DOSAGEM E ADMINISTRAÇÃO ]. As alterações nos valores laboratoriais selecionados durante a terapia estão descritas na Tabela 8. A maioria das alterações nos valores laboratoriais no ensaio PegIntron/REBETOL 200mg com pediatria foi leve ou moderada.
Hemoglobina
Os níveis de hemoglobina diminuíram para menos de 11 g/dL em cerca de 30% dos indivíduos no Estudo 2. No Estudo 3, 47% dos indivíduos que receberam WBD REBETOL 200mg e 33% em dose fixa de REBETOL 200mg tiveram reduções nos níveis de hemoglobina inferiores a 11 g /dl. Reduções na hemoglobina para menos de 9 g/dL ocorreram com mais frequência em indivíduos que receberam WBD em comparação com a dosagem plana (4% e 2%, respectivamente). No Estudo 2, a modificação da dose foi necessária em 9% e 13% dos indivíduos nos grupos PegIntron/REBETOL 200mg e INTRON A/REBETOL 200mg. No Estudo 4, os indivíduos que receberam PegIntron (1,5 mcg/kg)/REBETOL tiveram reduções nos níveis de hemoglobina entre 8,5 e menos de 10 g/dL (28%) e menos de 8,5 g/dL (3%), enquanto que em pacientes recebendo Pegasys 180 mcg/Copegus, essas diminuições ocorreram em 26% e 4% dos indivíduos, respectivamente. Os níveis de hemoglobina tornaram-se estáveis nas semanas de tratamento 4-6, em média. O padrão típico observado foi uma diminuição nos níveis de hemoglobina na semana 4 do tratamento, seguida de estabilização e um platô, que se manteve até o final do tratamento. No ensaio de monoterapia com PegIntron, as diminuições de hemoglobina foram geralmente leves e raramente foram necessárias modificações de dose [ver DOSAGEM E ADMINISTRAÇÃO ].
Neutrófilos
Foram observadas diminuições na contagem de neutrófilos na maioria dos indivíduos adultos tratados com terapia combinada com REBETOL 200mg no Estudo 2 (85%) e INTRON A/REBETOL (60%). Neutropenia grave potencialmente fatal (menos de 0,5 x 109/L) ocorreu em 2% dos indivíduos tratados com INTRON A/REBETOL 200mg e em aproximadamente 4% dos indivíduos tratados com PegIntron/REBETOL 200mg no Estudo 2. Dezoito por cento dos indivíduos recebendo PegIntron/REBETOL no Estudo 2 exigiu modificação da dosagem de interferon. Poucos indivíduos (menos de 1%) necessitaram de descontinuação permanente do tratamento. A contagem de neutrófilos geralmente retornou aos níveis pré-tratamento 4 semanas após a interrupção da terapia [ver DOSAGEM E ADMINISTRAÇÃO ].
Plaquetas
contagem de plaquetas diminuiu para menos de 100.000/mm³ em aproximadamente 20% dos indivíduos tratados com PegIntron isolado ou com REBETOL 200mg e em 6% dos indivíduos adultos tratados com INTRON A/REBETOL. Diminuições graves na contagem de plaquetas (menos de 50.000/mm³) ocorrem em menos de 4% dos indivíduos adultos. Os pacientes podem necessitar de descontinuação ou modificação da dose como resultado da diminuição das plaquetas [ver DOSAGEM E ADMINISTRAÇÃO ]. No Estudo 2, 1% ou 3% dos indivíduos necessitaram de modificação da dose de INTRON A ou PegIntron, respectivamente. A contagem de plaquetas geralmente retornou aos níveis pré-tratamento 4 semanas após a interrupção da terapia.
A função da tireóide
desenvolvimento de anormalidades do TSH, com ou sem manifestações clínicas, está associado às terapias com interferon. No Estudo 2, distúrbios da tireoide clinicamente aparentes ocorreram em indivíduos tratados com INTRON A ou PegIntron (com ou sem REBETOL) com incidência semelhante (5% para hipotireoidismo e 3% para hipertireoidismo). Os indivíduos desenvolveram novas anormalidades de TSH durante o tratamento e durante o período de acompanhamento. No final do período de acompanhamento, 7% dos indivíduos ainda apresentavam valores anormais de TSH.
Bilirrubina e Ácido Úrico
No Estudo 2, 10 a 14% dos indivíduos desenvolveram hiperbilirrubinemia e 33 a 38% desenvolveram hiperuricemia em associação com hemólise. Seis indivíduos desenvolveram gota leve a moderada.
Experiência em Ensaios Clínicos - Terapia Combinada REBETOL/INTRON A
Assuntos adultos
Em ensaios clínicos, 19% e 6% dos indivíduos previamente não tratados e recidivantes, respectivamente, descontinuaram a terapia devido a reações adversas nos braços de combinação em comparação com 13% e 3% nos braços de interferon. As reações adversas relacionadas ao tratamento selecionadas que ocorreram nos estudos norte-americanos com incidência maior ou igual a 5% são fornecidas por grupo de tratamento (ver Tabela 9). Em geral, as reações adversas relacionadas ao tratamento selecionadas foram relatadas com menor incidência nos estudos internacionais em comparação com os estudos norte-americanos, com exceção de astenia, sintomas semelhantes aos da gripe, nervosismo e prurido.
Sujeitos Pediátricos
Em ensaios clínicos de 118 indivíduos pediátricos de 3 a 16 anos de idade, 6% descontinuaram a terapia devido a reações adversas. Modificações de dose foram necessárias em 30% dos indivíduos, mais comumente para anemia e neutropenia. Em geral, o perfil de reações adversas na população pediátrica foi semelhante ao observado em adultos. Distúrbios no local da injeção, febre, anorexia, vômitos e labilidade emocional ocorreram com mais frequência em pacientes pediátricos em comparação com adultos. Por outro lado, os indivíduos pediátricos experimentaram menos fadiga, dispepsia, artralgia, insônia, irritabilidade, diminuição da concentração, dispneia e prurido em comparação com indivíduos adultos. As reações adversas relacionadas ao tratamento selecionadas que ocorreram com incidência maior ou igual a 5% entre todos os pacientes pediátricos que receberam a dose recomendada de terapia combinada REBETOL/INTRON A são fornecidas na Tabela 9.
Durante um curso de terapia de 48 semanas houve uma diminuição na taxa de crescimento linear (diminuição média de atribuição de percentil de 7%) e uma diminuição na taxa de ganho de peso (diminuição média de atribuição de percentil de 9%). Uma reversão geral dessas tendências foi observada durante o período pós-tratamento de 24 semanas. Dados de longo prazo em um número limitado de pacientes, no entanto, sugerem que a terapia combinada pode induzir uma inibição do crescimento que resulta na redução da altura final do adulto em alguns pacientes [ver AVISOS E PRECAUÇÕES ].
Valores de laboratório
As alterações nos valores hematológicos selecionados (hemoglobina, glóbulos brancos, neutrófilos e plaquetas) durante a terapia são descritas abaixo (consulte a Tabela 10).
Hemoglobina. diminuição da hemoglobina entre os indivíduos que receberam terapia com REBETOL começou na Semana 1, com estabilização na Semana 4. Em indivíduos não tratados previamente tratados por 48 semanas, a redução máxima média da linha de base foi de 3,1 g/dL no estudo americano e 2,9 g/dL no internacional tentativas. Em indivíduos com recaída, a redução máxima média da linha de base foi de 2,8 g/dL no estudo americano e 2,6 g/dL no estudo internacional. Os valores de hemoglobina retornaram aos níveis pré-tratamento dentro de 4 a 8 semanas após a interrupção da terapia na maioria dos indivíduos.
Bilirrubina e Ácido Úrico. Aumentos na bilirrubina e no ácido úrico, associados à hemólise, foram observados em ensaios clínicos. A maioria eram alterações bioquímicas moderadas e foram revertidas dentro de 4 semanas após a descontinuação do tratamento. Essa observação ocorreu com mais frequência em indivíduos com diagnóstico prévio de síndrome de Gilbert. Isso não foi associado a disfunção hepática ou morbidade clínica.
Experiências pós-marketing
As seguintes reações adversas foram identificadas e relatadas durante o uso pós-aprovação de REBETOL em combinação com INTRON A ou PegIntron. Como essas reações são relatadas voluntariamente por uma população de tamanho incerto, nem sempre é possível estimar com segurança sua frequência ou estabelecer uma relação causal com a exposição ao medicamento.
Distúrbios do Sangue e do Sistema Linfático
Aplasia pura de eritrócitos, anemia aplástica
Distúrbios do ouvido e labirinto
Distúrbio auditivo, vertigem
Distúrbios Respiratórios, Torácicos e Mediastinais
Hipertensão pulmonar
Distúrbios oculares
Descolamento seroso de retina
Distúrbios Endócrinos
Diabetes
INTERAÇÕES MEDICAMENTOSAS
Didanosina
exposição à didanosina ou seu metabólito ativo (didesoxiadenosina 5'-trifosfato) aumenta quando a didanosina é coadministrada com ribavirina, o que pode causar ou agravar toxicidades clínicas; portanto, a coadministração de REBETOL 200mg cápsulas ou solução oral e didanosina é contraindicada. Relatos de insuficiência hepática fatal, bem como neuropatia periférica, pancreatite e hiperlactatemia sintomática/acidose lática foram relatados em ensaios clínicos.
Análogos de Nucleosídeos
Descompensação hepática (algum fatal) ocorreu em pacientes cirróticos coinfectados com HIV/HCV recebendo terapia antirretroviral combinada para HIV e interferon alfa e ribavirina. A adição de tratamento com interferons alfa isolados ou em combinação com ribavirina pode aumentar o risco nesta população de pacientes. Pacientes recebendo interferon com ribavirina e inibidores nucleosídeos da transcriptase reversa (NRTIs) devem ser monitorados de perto para toxicidades associadas ao tratamento, especialmente descompensação hepática e anemia. A descontinuação de NRTIs deve ser considerada clinicamente apropriada (ver rotulagem para produto NRTI individual ). A redução da dose ou a descontinuação de interferon, ribavirina ou ambos também devem ser consideradas se forem observadas toxicidades clínicas agravadas, incluindo descompensação hepática (por exemplo, Child-Pugh maior que 6).
ribavirina pode antagonizar a atividade antiviral da cultura de células da estavudina e da zidovudina contra o HIV. A ribavirina demonstrou em cultura de células inibir a fosforilação da lamivudina, estavudina e zidovudina, o que pode levar à diminuição da atividade antirretroviral. No entanto, em um estudo com outro interferon peguilado em combinação com ribavirina, nenhuma interação farmacocinética (por exemplo, concentrações plasmáticas ou concentrações de metabólito ativo trifosforilado intracelular) ou farmacodinâmica (por exemplo, perda de supressão virológica de HIV/HCV) foi observada quando ribavirina e lamivudina (n =18), estavudina (n=10) ou zidovudina (n=6) foram coadministrados como parte de um regime multimedicamentoso em indivíduos coinfectados com HIV/HCV. Portanto, o uso concomitante de ribavirina com qualquer um desses medicamentos deve ser usado com cautela.
Drogas Metabolizadas pelo Citocromo P-450
Resultados de estudos in vitro usando preparações de microssomas hepáticos de humanos e ratos indicaram pouco ou nenhum metabolismo da ribavirina mediado pela enzima do citocromo P-450, com potencial mínimo para interações medicamentosas baseadas na enzima P-450.
Não foram observadas interações farmacocinéticas entre INTRON A e REBETOL 200 mg cápsulas em um estudo farmacocinético de dose múltipla.
Azatioprina
Foi relatado que o uso de ribavirina para o tratamento da hepatite C crônica em pacientes recebendo azatioprina induz pancitopenia grave e pode aumentar o risco de mielotoxicidade relacionada à azatioprina. A inosina monofosfato desidrogenase (IMDH) é necessária para uma das vias metabólicas da azatioprina. A ribavirina é conhecida por inibir a IMDH, levando ao acúmulo de um metabólito da azatioprina, monofosfato de 6-metiltioinosina (6-MTITP), que está associado à mielotoxicidade (neutropenia, trombocitopenia e anemia). Os pacientes que recebem azatioprina com ribavirina devem ter hemogramas completos, incluindo contagem de plaquetas, monitorados semanalmente no primeiro mês, duas vezes ao mês no segundo e terceiro meses de tratamento e, em seguida, mensalmente ou com mais frequência, se a dosagem ou outras alterações na terapia forem necessárias. AVISOS E PRECAUÇÕES ].
AVISOS
Incluído como parte do PRECAUÇÕES seção.
PRECAUÇÕES
Gravidez
REBETOL 200mg cápsulas e solução oral podem causar defeitos congênitos e morte do feto. A terapia com REBETOL não deve ser iniciada até que um relatório de um teste de gravidez negativo tenha sido obtido imediatamente antes do início planejado da terapia. As pacientes devem usar pelo menos duas formas de contracepção e fazer testes de gravidez mensais durante o tratamento e durante o período de 6 meses após a interrupção do tratamento. Extremo cuidado deve ser tomado para evitar a gravidez em pacientes do sexo feminino e em parceiras de pacientes do sexo masculino. REBETOL 200mg demonstrou efeitos teratogénicos e embriocidas significativos em todas as espécies animais em que foram realizados estudos adequados. Esses efeitos ocorreram em doses como inferior a um vigésimo da dose humana recomendada de ribavirina. A terapia com REBETOL 200mg não deve ser iniciada até que seja relatado um teste de gravidez negativo foi obtido imediatamente antes do início planejado da terapia [ver AVISO EM CAIXA , CONTRA-INDICAÇÕES , Uso em populações específicas , e INFORMAÇÃO DO PACIENTE ].
Anemia
toxicidade primária da ribavirina é a anemia hemolítica, que foi observada em aproximadamente 10% dos indivíduos tratados com REBETOL/INTRON A em ensaios clínicos. A anemia associada às cápsulas de REBETOL ocorre dentro de 1 a 2 semanas após o início da terapia. Como a queda inicial da hemoglobina pode ser significativa, recomenda-se que a hemoglobina ou o hematócrito sejam obtidos antes do início do tratamento e na Semana 2 e Semana 4 da terapia, ou com mais frequência, se clinicamente indicado. Os pacientes devem então ser acompanhados conforme clinicamente apropriado [ver DOSAGEM E ADMINISTRAÇÃO ].
Infartos do miocárdio fatais e não fatais foram relatados em pacientes com anemia causada por REBETOL. Os pacientes devem ser avaliados para doença cardíaca subjacente antes do início da terapia com ribavirina. Pacientes com doença cardíaca pré-existente devem realizar eletrocardiogramas antes do tratamento e devem ser monitorados adequadamente durante a terapia. Se houver qualquer deterioração do estado cardiovascular, a terapia deve ser suspensa ou descontinuada [ver DOSAGEM E ADMINISTRAÇÃO ]. Como a doença cardíaca pode ser agravada pela anemia induzida por drogas, pacientes com história de doença cardíaca significativa ou instável não devem usar REBETOL.
Pancreatite
A terapia com REBETOL e INTRON A ou PegIntron deve ser suspensa em pacientes com sinais e sintomas de pancreatite e descontinuada em pacientes com pancreatite confirmada.
Distúrbios Pulmonares
Sintomas pulmonares, incluindo dispneia, infiltrados pulmonares, pneumonite, hipertensão pulmonar e pneumonia, foram relatados durante a terapia com REBETOL com terapia combinada de interferon alfa; casos ocasionais de pneumonia fatal ocorreram. Além disso, foi relatada sarcoidose ou exacerbação da sarcoidose. Se houver evidência de infiltrados pulmonares ou comprometimento da função pulmonar, o paciente deve ser monitorado de perto e, se apropriado, a terapia combinada deve ser descontinuada.
Distúrbios Oftalmológicos
ribavirina é usada em terapia combinada com interferons alfa. Diminuição ou perda de visão, retinopatia incluindo edema macular, artéria ou veia retiniana, trombose, hemorragias retinianas e manchas algodonosas, neurite óptica, papiledema e descolamento seroso da retina são induzidas ou agravadas pelo tratamento com interferons alfa. Todos os pacientes devem receber um exame oftalmológico na linha de base. Pacientes com distúrbios oftalmológicos pré-existentes (por exemplo, retinopatia diabética ou hipertensiva) devem receber exames oftalmológicos periódicos durante a terapia combinada com o tratamento com interferon alfa. Qualquer paciente que desenvolva sintomas oculares deve receber um exame oftalmológico imediato e completo. A terapia combinada com interferons alfa deve ser descontinuada em pacientes que desenvolverem novos distúrbios oftalmológicos ou agravamento.
Testes laboratoriais
PegIntron em combinação com ribavirina pode causar graves diminuições nas contagens de neutrófilos e plaquetas e anormalidades hematológicas, endócrinas (por exemplo, TSH) e hepáticas.
Os pacientes em terapia de combinação PegIntron/REBETOL devem fazer exames de hematologia e química do sangue antes do início do tratamento e depois periodicamente. No ensaio clínico em adultos, contagens sanguíneas completas (incluindo contagem de hemoglobina, neutrófilos e plaquetas) e químicas (incluindo AST, ALT, bilirrubina e ácido úrico) foram medidas durante o período de tratamento nas semanas 2, 4, 8, 12 e em seguida, em intervalos de 6 semanas, ou mais freqüentemente se anormalidades se desenvolveram. Em indivíduos pediátricos, os mesmos parâmetros laboratoriais foram avaliados com avaliação adicional da hemoglobina na semana 6 de tratamento. Os níveis de TSH foram medidos a cada 12 semanas durante o período de tratamento. O HCV-RNA deve ser medido periodicamente durante o tratamento [ver DOSAGEM E ADMINISTRAÇÃO ].
Distúrbios Dentários e Periodontais
Distúrbios dentários e periodontais foram relatados em pacientes recebendo terapia combinada de ribavirina e interferon ou peginterferon. Além disso, a boca seca pode ter um efeito prejudicial nos dentes e nas membranas mucosas da boca durante o tratamento a longo prazo com a combinação de REBETOL e interferon alfa-2b peguilado ou não peguilado. Os pacientes devem escovar bem os dentes duas vezes ao dia e fazer exames odontológicos regulares. Se ocorrer vômito, eles devem ser aconselhados a enxaguar bem a boca depois.
Administração concomitante de azatioprina
Pancitopenia (diminuição acentuada dos glóbulos vermelhos, neutrófilos e plaquetas) e supressão da medula óssea foram relatados na literatura como ocorrendo dentro de 3 a 7 semanas após a administração concomitante de interferon peguilado/ribavirina e azatioprina. Neste número limitado de pacientes (n=8), a mielotoxicidade foi reversível dentro de 4 a 6 semanas após a retirada da terapia antiviral para HCV e azatioprina concomitante e não houve recorrência após a reintrodução de qualquer tratamento isolado. PegIntron, REBETOL e azatioprina devem ser descontinuados para pancitopenia, e interferon peguilado/ribavirina não deve ser reintroduzido com azatioprina concomitante [ver INTERAÇÕES MEDICAMENTOSAS ].
Impacto no Crescimento - Uso Pediátrico
Os dados sobre os efeitos de PegIntron e REBETOL no crescimento vêm de um estudo aberto em indivíduos de 3 a 17 anos de idade, no qual as alterações de peso e altura são comparadas com dados populacionais normativos dos EUA. Em geral, o ganho de peso e altura de pacientes pediátricos tratados com PegIntron e REBETOL 200mg fica atrás do previsto pelos dados normativos da população para toda a duração do tratamento. Velocidade de crescimento severamente inibida (menos de 3º percentil) foi observada em 70% dos indivíduos durante o tratamento. Após o tratamento, o crescimento rebote e o ganho de peso ocorreram na maioria dos indivíduos. No entanto, dados de acompanhamento de longo prazo em pacientes pediátricos indicam que PegIntron em terapia combinada com REBETOL pode induzir uma inibição do crescimento que resulta em redução da altura adulta em alguns pacientes [ver REAÇÕES ADVERSAS ].
Da mesma forma, um impacto no crescimento foi observado em indivíduos após tratamento com REBETOL 200mg e terapia combinada com INTRON A por um ano. Em um estudo de acompanhamento de longo prazo de um número limitado desses indivíduos, a terapia combinada resultou em redução da altura final do adulto em alguns indivíduos [ver REAÇÕES ADVERSAS ].
Salvaguardas de uso
Com base nos resultados dos ensaios clínicos, a monoterapia com ribavirina não é eficaz para o tratamento da infecção crônica pelo vírus da hepatite C; portanto, REBETOL 200mg cápsulas ou solução oral não deve ser usado isoladamente. A segurança e eficácia de REBETOL cápsulas e solução oral só foram estabelecidas quando usadas em conjunto com INTRON A ou PegIntron (não outros interferons) como terapia combinada.
segurança e eficácia da terapia com REBETOL/INTRON A e PegIntron para o tratamento de infecções por HIV, adenovírus, RSV, parainfluenza ou influenza não foram estabelecidas. REBETOL 200mg cápsulas não deve ser usado para essas indicações. A ribavirina para inalação tem rotulagem separada, que deve ser consultada se a terapia inalatória com ribavirina estiver sendo considerada.
Existem reações adversas significativas causadas pela terapia com REBETOL/INTRON A ou PegIntron, incluindo depressão grave e ideação suicida, anemia hemolítica, supressão da função da medula óssea, distúrbios autoimunes e infecciosos, disfunção pulmonar, pancreatite e diabetes. A ideação ou tentativa de suicídio ocorreu com maior frequência entre os pacientes pediátricos, principalmente adolescentes, em comparação aos pacientes adultos (2,4% versus 1%) durante o tratamento e acompanhamento fora da terapia. A rotulagem de INTRON A e PegIntron deve ser revisada em sua totalidade para obter informações de segurança adicionais antes do início do tratamento combinado.
Informações de Aconselhamento do Paciente
Aconselhe o paciente a ler a Rótulo do Paciente Aprovado pela FDA ( Guia de Medicação ).
Anemia
A experiência adversa mais comum que ocorre com REBETOL cápsulas é a anemia, que pode ser grave [ver AVISOS E PRECAUÇÕES e REAÇÕES ADVERSAS ]. Os pacientes devem ser avisados de que são necessárias avaliações laboratoriais antes do início da terapia e depois periodicamente [ver DOSAGEM E ADMINISTRAÇÃO ]. É aconselhável que os pacientes estejam bem hidratados, especialmente durante as fases iniciais do tratamento.
Gravidez
Os pacientes devem ser informados de que REBETOL 200mg cápsulas e solução oral podem causar defeitos congênitos e morte do feto. REBETOL 200mg não deve ser usado por mulheres grávidas ou por homens cujas parceiras estejam grávidas. Deve-se ter extremo cuidado para evitar a gravidez em pacientes do sexo feminino e em parceiras de pacientes do sexo masculino em uso de REBETOL. REBETOL não deve ser iniciado até que um relatório de teste de gravidez negativo tenha sido obtido imediatamente antes do início da terapia. Os pacientes devem realizar um teste de gravidez mensalmente durante a terapia e por 6 meses após a terapia.
Mulheres com potencial para engravidar devem ser aconselhadas sobre o uso de contracepção eficaz (duas formas confiáveis) antes de iniciar a terapia. Os pacientes (homens e mulheres) devem ser informados sobre os riscos teratogênicos/embriocidas e devem ser instruídos a praticar contracepção eficaz durante REBETOL 200mg e por 6 meses após a terapia. Os pacientes (homens e mulheres) devem ser aconselhados a notificar o médico imediatamente em caso de gravidez [ver CONTRA-INDICAÇÕES , AVISOS E PRECAUÇÕES , e Uso em populações específicas ].
Se ocorrer gravidez durante o tratamento ou durante 6 meses após a terapia, a paciente deve ser avisada do risco teratogênico da terapia com REBETOL 200mg para o feto. Pacientes, ou parceiros de pacientes, devem relatar imediatamente qualquer gravidez que ocorra durante o tratamento ou dentro de 6 meses após a interrupção do tratamento ao seu médico. Os prescritores devem relatar esses casos ligando para 1-800-593-2214.
Riscos versus benefícios
Pacientes recebendo REBETOL cápsulas devem ser informados sobre os benefícios e riscos associados ao tratamento, orientados quanto ao seu uso adequado e encaminhados ao paciente. Guia de Medicação . Os pacientes devem ser informados de que o efeito do tratamento da infecção por hepatite C na transmissão não é conhecido e que devem ser tomadas as precauções apropriadas para prevenir a transmissão do vírus da hepatite C.
Os pacientes devem ser informados sobre o que fazer caso percam uma dose de REBETOL; a dose esquecida deve ser tomada o mais rápido possível durante o mesmo dia. Os doentes não devem duplicar a dose seguinte. Os pacientes devem ser aconselhados a entrar em contato com seu médico se tiverem dúvidas.
Toxicologia não clínica
Carcinogênese, Mutagênese, Prejuízo da Fertilidade
Carcinogênese
ribavirina não causou um aumento em nenhum tipo de tumor quando administrada por 6 meses no modelo de camundongo transgênico deficiente em p53 em doses de até 300 mg/kg (equivalente humano estimado de 25 mg/kg com base no ajuste da área de superfície corporal para um adulto de 60 kg ; aproximadamente 1,9 vezes a dose diária humana máxima recomendada). A ribavirina não foi carcinogênica quando administrada por 2 anos a ratos em doses de até 40 mg/kg (equivalente humano estimado de 5,71 mg/kg com base no ajuste da área de superfície corporal para um adulto de 60 kg).
Mutagênese
ribavirina demonstrou incidências aumentadas de mutação e transformação celular em vários ensaios de genotoxicidade. A ribavirina foi ativa no Ensaio de Transformação Celular In Vitro Balb/3T3. A atividade mutagênica foi observada no ensaio de linfoma de camundongo e em doses de 20 a 200 mg/kg (equivalente humano estimado de 1,67 a 16,7 mg/kg, com base no ajuste da área de superfície corporal para um adulto de 60 kg; 0,1 a 1 vezes o máximo dose humana recomendada de ribavirina em 24 horas) em um ensaio de micronúcleo em camundongo. Um ensaio letal dominante em ratos foi negativo, indicando que, se as mutações ocorreram em ratos, elas não foram transmitidas através de gametas masculinos.
Prejuízo da Fertilidade
ribavirina demonstrou efeitos embriocidas e teratogênicos significativos em doses bem abaixo da dose humana recomendada em todas as espécies animais nas quais foram realizados estudos adequados. Foram observadas malformações do crânio, palato, olho, mandíbula, membros, esqueleto e trato gastrointestinal. A incidência e a gravidade dos efeitos teratogênicos aumentaram com o aumento da dose do fármaco. A sobrevivência de fetos e descendentes foi reduzida. Em estudos convencionais de embriotoxicidade/teratogenicidade em ratos e coelhos, os níveis de dose sem efeito observados foram bem abaixo daqueles para uso clínico proposto (0,3 mg/kg/dia para ratos e coelhos; aproximadamente 0,06 vezes a dose humana recomendada de 24 horas de ribavirina). Nenhuma toxicidade materna ou efeitos na prole foram observados em um estudo de toxicidade peri/pós-natal em ratos com dose oral de até 1 mg/kg/dia (dose equivalente humana estimada de 0,17 mg/kg com base no ajuste da área de superfície corporal para um adulto de 60 kg ; aproximadamente 0,01 vezes a dose humana máxima recomendada de ribavirina em 24 horas) [ver CONTRA-INDICAÇÕES , e AVISOS E PRECAUÇÕES ].
Mulheres férteis e parceiros de mulheres férteis não devem receber REBETOL a menos que a paciente e seu parceiro estejam usando métodos contraceptivos eficazes (duas formas confiáveis). Com base em uma meia-vida de dose múltipla (t1/2) de ribavirina de 12 dias, a contracepção eficaz deve ser utilizada por 6 meses após a terapia (por exemplo, 15 meias-vidas de depuração da ribavirina).
REBETOL deve ser usado com cautela em homens férteis. Em estudos em camundongos para avaliar o curso de tempo e a reversibilidade da degeneração testicular induzida pela ribavirina em doses de 15 a 150 mg/kg/dia (equivalente humano estimado de 1,25 a 12,5 mg/kg/dia, com base no ajuste da área de superfície corporal para um adulto de 60 kg; 0,1-0,8 vezes a dose máxima humana de ribavirina em 24 horas administrada por 3 ou 6 meses, ocorreram anormalidades no esperma. Após a interrupção do tratamento, a recuperação essencialmente total da toxicidade testicular induzida pela ribavirina foi aparente em 1 ou 2 ciclos de espermatogênese.
Uso em populações específicas
Gravidez
Gravidez Categoria X
[Ver CONTRA-INDICAÇÕES , AVISOS E PRECAUÇÕES , e Toxicologia não clínica ].
Tratamento e Pós-tratamento:
Risco potencial para o feto
A ribavirina é conhecida por se acumular em componentes intracelulares de onde é eliminada muito lentamente. Não se sabe se a ribavirina contida no esperma exercerá um potencial efeito teratogênico na fertilização dos óvulos. Em um estudo em ratos, concluiu-se que a letalidade dominante não foi induzida pela ribavirina em doses de até 200 mg/kg por 5 dias (doses equivalentes humanos estimadas de 7,14 a 28,6 mg/kg, com base no ajuste da área de superfície corporal para 60 kg de adulto; até 1,7 vezes a dose humana máxima recomendada de ribavirina). No entanto, devido aos potenciais efeitos teratogénicos humanos da ribavirina, os doentes do sexo masculino devem ser aconselhados a tomar todas as precauções para evitar o risco de gravidez para as suas parceiras.
Mulheres com potencial para engravidar não devem receber REBETOL a menos que estejam usando métodos contraceptivos eficazes (duas formas confiáveis) durante o período de terapia. Além disso, a contracepção eficaz deve ser utilizada por 6 meses após a terapia com base em uma meia-vida de dose múltipla (t1/2) de ribavirina de 12 dias.
Pacientes do sexo masculino e suas parceiras devem praticar contracepção eficaz (duas formas confiáveis) durante o tratamento com REBETOL e durante o período de 6 meses após a terapia (por exemplo, 15 meias-vidas para a depuração da ribavirina do corpo).
Um Registro de Gravidez Ribavirina foi estabelecido para monitorar os resultados materno-fetais de gestações em pacientes do sexo feminino e parceiras de pacientes do sexo masculino expostos à ribavirina durante o tratamento e por 6 meses após a interrupção do tratamento. Médicos e pacientes são encorajados a relatar tais casos ligando para 1-800-593-2214.
Mães que amamentam
Não se sabe se o produto REBETOL é excretado no leite humano. Devido ao potencial de reações adversas graves do medicamento em lactentes, deve-se decidir entre descontinuar a amamentação ou atrasar ou descontinuar REBETOL.
Uso Pediátrico
A segurança e eficácia de REBETOL 200mg em combinação com PegIntron não foi estabelecida em pacientes pediátricos com idade inferior a 3 anos. Para o tratamento com REBETOL/INTRON A, evidências de progressão da doença, como inflamação hepática e fibrose, bem como fatores prognósticos para resposta, genótipo do HCV e carga viral devem ser considerados ao decidir tratar um paciente pediátrico. Os benefícios do tratamento devem ser ponderados em relação aos achados de segurança observados.
Dados de acompanhamento de longo prazo em pacientes pediátricos indicam que REBETOL em combinação com PegIntron ou com INTRON A pode induzir uma inibição do crescimento que resulta em redução da altura em alguns pacientes [ver AVISOS E PRECAUÇÕES e REAÇÕES ADVERSAS ].
Ideação ou tentativas suicidas ocorreram com mais frequência entre pacientes pediátricos, principalmente adolescentes, em comparação com pacientes adultos (2,4% vs. 1%) durante o tratamento e acompanhamento fora da terapia [Vejo AVISOS E PRECAUÇÕES ]. Tal como nos doentes adultos, os doentes pediátricos apresentaram outras reações adversas psiquiátricas (por exemplo, depressão, labilidade emocional, sonolência), anemia e neutropenia [ver AVISOS E PRECAUÇÕES ].
Uso Geriátrico
Os ensaios clínicos da terapia com REBETOL/INTRON A ou PegIntron não incluíram um número suficiente de indivíduos com 65 anos ou mais para determinar se eles respondem de forma diferente dos indivíduos mais jovens.
REBETOL 200mg é conhecido por ser substancialmente excretado pelos rins, e o risco de reações tóxicas a esta droga pode ser maior em pacientes com função renal comprometida. Como os pacientes idosos geralmente apresentam função renal diminuída, deve-se ter cuidado na seleção da dose. A função renal deve ser monitorada e os ajustes de dosagem devem ser feitos de acordo. REBETOL 200mg não deve ser usado em pacientes com clearance de creatinina menor que 50 mL/min [ver CONTRA-INDICAÇÕES ].
Em geral, REBETOL 200mg cápsulas deve ser administrado a pacientes idosos com cautela, começando na extremidade inferior da faixa de dosagem, refletindo a maior frequência de diminuição da função hepática e cardíaca e de doença concomitante ou outra terapia medicamentosa. Em ensaios clínicos, indivíduos idosos tiveram uma frequência maior de anemia (67%) do que pacientes mais jovens (28%) [ver AVISOS E PRECAUÇÕES ].
Destinatários de Transplante de Órgãos
segurança e eficácia de INTRON A e PegIntron isoladamente ou em combinação com REBETOL para o tratamento da hepatite C em receptores de transplante de fígado ou outros órgãos não foram estabelecidas. Em uma pequena (n=16) experiência de caso não controlado em um único centro, a insuficiência renal em receptores de aloenxerto renal recebendo terapia combinada de interferon alfa e ribavirina foi mais frequente do que o esperado a partir da experiência anterior do centro com receptores de aloenxerto renal que não receberam terapia combinada. A relação da insuficiência renal com a rejeição do aloenxerto renal não é clara.
Co-infecção HIV ou HBV
A segurança e eficácia de PegIntron/REBETOL 200mg e INTRON A/REBETOL para o tratamento de pacientes com HCV coinfectados com HIV ou HBV não foram estabelecidas.
SOBREDOSAGEM
experiência com sobredosagem é limitada. Ingestão aguda de até 20 g de cápsulas de REBETOL, ingestão de INTRON A de até 120 milhões de unidades e doses subcutâneas de INTRON A até 10 vezes as doses recomendadas foram relatadas. Os efeitos primários observados são o aumento da incidência e gravidade das reações adversas relacionadas ao uso terapêutico de INTRON A e REBETOL. No entanto, anormalidades das enzimas hepáticas, insuficiência renal, hemorragia e infarto do miocárdio foram relatados com a administração de doses subcutâneas únicas de INTRON A que excedem as recomendações de dosagem.
Não há antídoto específico para a superdosagem de INTRON A ou REBETOL 200mg, e a hemodiálise e a diálise peritoneal não são eficazes para o tratamento da superdosagem desses agentes.
CONTRA-INDICAÇÕES
A terapia combinada de REBETOL é contraindicada em:
- mulheres que estão grávidas. REBETOL 200mg pode causar dano fetal quando administrado a uma mulher grávida. REBETOL é contraindicado em mulheres grávidas ou que possam vir a engravidar. Se REBETOL for usado durante a gravidez, ou se a paciente engravidar enquanto estiver tomando REBETOL 200mg, a paciente deve ser informada do risco potencial para o feto [ver AVISOS E PRECAUÇÕES , Uso em populações específicas , e INFORMAÇÃO DO PACIENTE ]
- homens cujas parceiras estão grávidas
- pacientes com reações de hipersensibilidade conhecidas, como síndrome de Stevens-Johnson, necrólise epidérmica tóxica e eritema multiforme à ribavirina ou a qualquer componente do produto
- pacientes com hepatite autoimune
- pacientes com hemoglobinopatias (por exemplo, talassemia maior, anemia falciforme)
- pacientes com clearance de creatinina menor que 50 mL/min. [Vejo Uso em populações específicas e FARMACOLOGIA CLÍNICA ]
- coadministração de REBETOL e didanosina é contraindicada porque a exposição ao metabólito ativo da didanosina (didesoxiadenosina 5'-trifosfato) é aumentada. Insuficiência hepática fatal, bem como neuropatia periférica, pancreatite e hiperlactatemia/acidose lática sintomática foram relatadas em pacientes recebendo didanosina em combinação com ribavirina [ver INTERAÇÕES MEDICAMENTOSAS ].
FARMACOLOGIA CLÍNICA
Mecanismo de ação
A ribavirina é um agente antiviral [ver Microbiologia ].
Farmacocinética
As propriedades farmacocinéticas de dose única e múltipla em adultos estão resumidas na Tabela 11. A ribavirina foi rápida e extensivamente absorvida após administração oral. No entanto, devido ao metabolismo de primeira passagem, a biodisponibilidade absoluta foi em média de 64% (44%). Houve uma relação linear entre a dose e AUCtf (AUC do tempo zero até a última concentração mensurável) após doses únicas de 200 a 1200 mg de ribavirina. A relação entre dose e Cmax foi curvilínea, tendendo a assíntotas acima de doses únicas de 400 a 600 mg.
Após administração oral múltipla, com base na AUC12h, foi observada uma acumulação de 6 vezes de ribavirina no plasma. Após administração oral de 600 mg duas vezes ao dia, o estado de equilíbrio foi alcançado em aproximadamente 4 semanas, com concentrações plasmáticas médias no estado de equilíbrio de 2.200 ng/mL (37%). Após a descontinuação da dosagem, a meia-vida média foi de 298 (30%) horas, o que provavelmente reflete a eliminação lenta dos compartimentos não plasmáticos.
Efeito do Antiácido na Absorção da Ribavirina
A coadministração de REBETOL cápsulas com um antiácido contendo magnésio, alumínio e simeticona resultou em uma diminuição de 14% na AUCtf média da ribavirina. A relevância clínica dos resultados deste estudo de dose única é desconhecida.
Distribuição de tecidos
transporte de ribavirina para compartimentos não plasmáticos foi mais extensivamente estudado em glóbulos vermelhos, e foi identificado como sendo principalmente por meio de um transportador de nucleosídeo equilibrado do tipo es. Este tipo de transportador está presente em praticamente todos os tipos de células e pode ser responsável pelo extenso volume de distribuição. A ribavirina não se liga às proteínas plasmáticas.
Metabolismo e Excreção
A ribavirina tem duas vias de metabolismo: (i) uma via de fosforilação reversível em células nucleadas; e (ii) uma via de degradação envolvendo desribosilação e hidrólise de amida para produzir um metabólito de ácido triazol carboxílico. A ribavirina e seus metabólitos triazol carboxamida e triazol carboxílico são excretados por via renal. Após administração oral de 600 mg de 14C-ribavirina, aproximadamente 61% e 12% da radioatividade foi eliminada na urina e nas fezes, respectivamente, em 336 horas. A ribavirina inalterada representou 17% da dose administrada.
Populações Especiais
Disfunção Renal
farmacocinética da ribavirina foi avaliada após a administração de uma dose oral única (400 mg) de ribavirina a indivíduos não infectados pelo HCV com graus variados de disfunção renal. O valor médio de AUCtf foi três vezes maior em indivíduos com valores de depuração de creatinina entre 10 a 30 mL/min quando comparados a indivíduos controle (depuração de creatinina maior que 90 mL/min). Em indivíduos com valores de depuração de creatinina entre 30 a 60 mL/min, a AUCtf foi duas vezes maior quando comparado aos indivíduos controle. O aumento da AUCtf parece ser devido à redução da depuração renal e não renal nesses indivíduos. Os ensaios de eficácia da Fase 3 incluíram indivíduos com valores de depuração de creatinina superiores a 50 mL/min. A farmacocinética de doses múltiplas da ribavirina não pode ser prevista com precisão em pacientes com disfunção renal. A ribavirina não é efetivamente removida por hemodiálise.
Pacientes com clearance de creatinina menor que 50 mL/min não devem ser tratados com REBETOL [ver CONTRA-INDICAÇÕES ].
Disfunção Hepática
efeito da disfunção hepática foi avaliado após uma dose oral única de ribavirina (600 mg). Os valores médios de AUCtf não foram significativamente diferentes em indivíduos com disfunção hepática leve, moderada ou grave (Classificação de Child-Pugh A, B ou C) quando comparados a indivíduos controle. No entanto, os valores médios de Cmax aumentaram com a gravidade da disfunção hepática e foram duas vezes maiores em indivíduos com disfunção hepática grave quando comparados aos indivíduos controle.
Pacientes idosos
Não foram realizadas avaliações farmacocinéticas em indivíduos idosos.
Gênero
Não houve diferenças farmacocinéticas clinicamente significativas observadas em um estudo de dose única de 18 indivíduos do sexo masculino e 18 do sexo feminino.
Pacientes pediátricos
As propriedades farmacocinéticas de dose múltipla de REBETOL 200mg cápsulas e INTRON A em pacientes pediátricos com hepatite C crônica entre 5 e 16 anos de idade estão resumidas na Tabela 12. A farmacocinética de REBETOL 200mg e INTRON A (dose normalizada) é semelhante em adultos e assuntos pediátricos.
As características farmacocinéticas completas de REBETOL solução oral não foram determinadas em pacientes pediátricos. Os valores de Cmin da ribavirina foram semelhantes após a administração de REBETOL solução oral ou REBETOL 200 mg cápsulas durante 48 semanas de terapia em pacientes pediátricos (3 a 16 anos de idade).
Foi realizado um ensaio clínico em pacientes pediátricos com hepatite C crônica entre 3 e 17 anos de idade, no qual a farmacocinética de PegIntron e REBETOL (cápsulas e solução oral) foi avaliada. Em indivíduos pediátricos que receberam a dosagem ajustada à área de superfície corporal de PegIntron a 60 mcg/m²/semana, a estimativa da razão log transformada da exposição durante o intervalo de dosagem foi prevista para ser 58% [IC 90%: 141%, 177%] maior do que observada em adultos recebendo 1,5 mcg/kg/semana. A farmacocinética de REBETOL (dose normalizada) neste estudo foi semelhante à relatada em um estudo anterior de REBETOL 200mg em combinação com INTRON A em pacientes pediátricos e em adultos.
Efeito dos Alimentos na Absorção da Ribavirina
Tanto a AUCtf quanto a Cmax aumentaram 70% quando as cápsulas de REBETOL foram administradas com uma refeição rica em gordura (841 kcal, 53,8 g de gordura, 31,6 g de proteína e 57,4 g de carboidrato) em um estudo farmacocinético de dose única [ver DOSAGEM E ADMINISTRAÇÃO ].
Microbiologia
Mecanismo de ação
O mecanismo pelo qual a ribavirina contribui para sua eficácia antiviral na clínica não é totalmente compreendido. A ribavirina tem atividade antiviral direta na cultura de tecidos contra muitos vírus de RNA. A ribavirina aumenta a frequência de mutação nos genomas de vários vírus e o trifosfato de ribavirina inibe a polimerase do HCV em uma reação bioquímica.
Atividade antiviral em cultura celular
atividade antiviral da ribavirina no replicão do HCV não é bem compreendida e não foi definida devido à toxicidade celular da ribavirina. A atividade antiviral direta foi observada em cultura de tecidos de outros vírus de RNA. A atividade anti-HCV do interferon foi demonstrada em células contendo HCV-RNS autorreplicantes (células replicon de HCV) ou infecção por HCV.
Resistência
Os genótipos de HCV mostram uma grande variabilidade na sua resposta à terapia com interferon/ribavirina recombinante peguilado humano. Não foram identificadas alterações genéticas associadas à resposta variável.
Resistência cruzada
Não há relato de resistência cruzada entre interferons peguilados/não peguilados e ribavirina.
Toxicologia e Farmacologia Animal
Estudos de longo prazo em camundongos e ratos [18 a 24 meses; doses de 20 a 75 e 10 a 40 mg/kg/dia, respectivamente (doses equivalentes humanas estimadas de 1,67 a 6,25 e 1,43 a 5,71 mg/kg/dia, respectivamente, com base no ajuste da área de superfície corporal para um adulto de 60 kg; aproximadamente 0,1 a 0,4 vezes a dose máxima humana de ribavirina em 24 horas)] demonstraram uma relação entre a exposição crônica à ribavirina e o aumento da incidência de lesões vasculares (hemorragias microscópicas) em camundongos. Em ratos, a degeneração da retina ocorreu em controles, mas a incidência foi aumentada em ratos tratados com ribavir.
Em um estudo no qual filhotes de ratos receberam doses pós-natal de ribavirina em doses de 10, 25 e 50 mg/kg/dia, as mortes relacionadas à droga ocorreram em 50 mg/kg (nas concentrações plasmáticas de filhotes de ratos abaixo das concentrações plasmáticas humanas no nível humano). dose terapêutica) entre os Dias 13 e 48 do estudo. Filhotes de ratos dosados dos Dias 7 a 63 pós-natais demonstraram uma diminuição menor relacionada à dose no crescimento geral em todas as doses, que se manifestou posteriormente como ligeiras diminuições no peso corporal, comprimento coroa-nádega, e comprimento ósseo. Esses efeitos mostraram evidência de reversibilidade, e nenhum efeito histopatológico no osso foi observado. Não foram observados efeitos da ribavirina em relação ao desenvolvimento neurocomportamental ou reprodutivo.
Estudos clínicos
O Estudo Clínico 1 avaliou a monoterapia com PegIntron. Ver Rotulagem PegIntron para obter informações sobre este estudo.
Terapia combinada REBETOL/PegIntron
Assuntos adultos
Estudo 2
Um estudo randomizado comparou o tratamento com dois regimes de PegIntron/REBETOL 200mg [PegIntron 1,5 mcg/kg por via subcutânea uma vez por semana/REBETOL 800 mg por via oral diariamente (em doses divididas); PegIntron 1,5 mcg/kg por via subcutânea uma vez por semana durante 4 semanas, depois 0,5 mcg/kg por via subcutânea uma vez por semana durante 44 semanas/REBETOL 1000 ou 1200 mg por via oral diariamente (em doses divididas)] com INTRON A [3 MUI por via subcutânea três vezes por semana/REBETOL 1000 ou 1.200 mg por via oral diariamente (em doses divididas)] em 1.530 adultos com hepatite C crônica. Indivíduos sem tratamento prévio com interferon foram tratados por 48 semanas e acompanhados por 24 semanas após o tratamento. Indivíduos elegíveis tinham doença hepática compensada, HCV-RNA detectável, ALT elevada e histopatologia hepática consistente com hepatite crônica.
resposta ao tratamento foi definida como HCV-RNA indetectável 24 semanas após o tratamento (ver Tabela 13). A taxa de resposta à dose de PegIntron 1,5 mcg/kg e ribavirina 800 mg foi maior do que a taxa de resposta a INTRON A/REBETOL (consulte a Tabela 13). A taxa de resposta a PegIntron 1,5→0,5 mcg/kg/REBETOL 200mg foi essencialmente a mesma como a resposta a INTRON A/REBETOL (dados não mostrados).
Indivíduos com genótipo viral 1, independentemente da carga viral, tiveram uma taxa de resposta mais baixa ao PegIntron (1,5 mcg/kg)/REBETOL (800 mg) em comparação com indivíduos com outros genótipos virais. Indivíduos com ambos os fatores de mau prognóstico (genótipo 1 e alta carga viral) tiveram uma taxa de resposta de 30% (78/256) em comparação com uma taxa de resposta de 29% (71/247) com a terapia combinada INTRON A/REBETOL.
Indivíduos com menor peso corporal tenderam a ter maiores taxas de reações adversas [ver REAÇÕES ADVERSAS e taxas de resposta mais altas do que indivíduos com pesos corporais mais altos. As diferenças nas taxas de resposta entre os braços de tratamento não variaram substancialmente com o peso corporal.
As taxas de resposta ao tratamento com a terapia combinada PegIntron/REBETOL foram de 49% em homens e 56% em mulheres. As taxas de resposta foram menores em indivíduos afro-americanos e hispânicos e maiores em asiáticos em comparação com caucasianos. Embora os afro-americanos tivessem uma proporção maior de fatores de mau prognóstico em comparação com os caucasianos, o número de não caucasianos estudados (11% do total) foi insuficiente para permitir conclusões significativas sobre as diferenças nas taxas de resposta após o ajuste para fatores de prognóstico neste estudo.
Biópsias hepáticas foram obtidas antes e após o tratamento em 68% dos indivíduos. Em comparação com a linha de base, observou-se que aproximadamente dois terços dos indivíduos em todos os grupos de tratamento tiveram uma redução modesta na inflamação.
Estudo 3
Em um grande estudo de base comunitária nos Estados Unidos, 4.913 indivíduos com hepatite C crônica foram randomizados para receber PegIntron 1,5 mcg/kg por via subcutânea uma vez por semana em combinação com uma dose de REBETOL 200 mg de 800 a 1400 mg (dosagem baseada no peso [WBD]) ou 800 mg (plano) por via oral diariamente (em doses divididas) por 24 ou 48 semanas com base no genótipo. A resposta ao tratamento foi definida como HCV-RNA indetectável (com base em um ensaio com um limite inferior de detecção de 125 UI/mL) em 24 semanas após o tratamento.
tratamento com PegIntron 1,5 mcg/kg e REBETOL 800 a 1400 mg resultou em uma resposta virológica sustentada mais alta em comparação com PegIntron em combinação com uma dose diária fixa de 800 mg de REBETOL. Indivíduos com peso superior a 105 kg obtiveram o maior benefício com WBD, embora também tenha sido observado um benefício modesto em indivíduos com peso superior a 85 a 105 kg (ver Tabela 14). O benefício do WBD em indivíduos com peso superior a 85 kg foi observado com os genótipos 1-3 do HCV. Dados insuficientes estavam disponíveis para chegar a conclusões sobre outros genótipos. O uso de WBD resultou em um aumento da incidência de anemia [ver REAÇÕES ADVERSAS ].
Um total de 1.552 indivíduos com peso superior a 65 kg no Estudo 3 tinham genótipo 2 ou 3 e foram randomizados para 24 ou 48 semanas de terapia. Nenhum benefício adicional foi observado com a duração mais longa do tratamento.
Estudo 4
Um grande estudo randomizado comparou a segurança e eficácia do tratamento por 48 semanas com dois regimes de PegIntron/REBETOL 200mg [PegIntron 1,5 mcg/kg e 1 mcg/kg subcutaneamente uma vez por semana, ambos em combinação com REBETOL 800 a 1400 mg PO diariamente (em dois doses)] e Pegasys 180 mcg por via subcutânea uma vez por semana em combinação com Copegus 1.000 a 1.200 mg VO diariamente (em duas doses divididas) em 3.070 adultos virgens de tratamento com genótipo 1 de hepatite C crônica. HCV-RNA ou redução maior ou igual a 2 log10 da linha de base) até o tratamento A Semana 12 foi o critério para descontinuação do tratamento. A RVS foi definida como HCV-RNA indetectável (ensaio Roche COBAS TaqMan, um limite inferior de quantificação de 27 UI/mL) às 24 semanas pós-tratamento (ver Tabela 15).
As taxas gerais de SVR foram semelhantes entre os três grupos de tratamento. Independentemente do grupo de tratamento, as taxas de RVS foram menores em indivíduos com fatores de mau prognóstico. Indivíduos com fatores de mau prognóstico randomizados para PegIntron (1,5 mcg/kg)/REBETOL ou Pegasys/Copegus, no entanto, alcançaram taxas de RVS mais altas em comparação com indivíduos semelhantes randomizados para PegIntron 1 mcg/kg/REBETOL. Para PegIntron 1,5 mcg/kg e dose de REBETOL, as taxas de RVS para indivíduos com e sem os seguintes fatores prognósticos foram as seguintes: cirrose (10% vs. 42%), níveis normais de ALT (32% vs. 42%), virais basais carga superior a 600.000 UI/mL (35% vs. 61%), 40 anos de idade ou mais (38% vs. 50%) e raça afro-americana (23% vs. 44%). Em indivíduos com HCV-RNA indetectável na Semana 12 de tratamento que receberam PegIntron (1,5 mcg/kg)/REBETOL 200mg, a taxa de RVS foi de 81% (328/407).
Estudo 5 - Terapia Combinada REBETOL/PegIntron em Falhas de Tratamentos Anteriores
Em um estudo não comparativo, 2.293 indivíduos com fibrose moderada a grave que falharam no tratamento anterior com a combinação interferon alfa/ribavirina foram tratados novamente com PegIntron, 1,5 mcg/kg por via subcutânea, uma vez por semana, em combinação com ribavirina ajustada ao peso. Indivíduos elegíveis incluíam não respondedores anteriores (indivíduos que eram HCV-RNA positivos ao final de um mínimo de 12 semanas de tratamento) e reincidentes anteriores (indivíduos que eram HCVRNA negativos ao final de um mínimo de 12 semanas de tratamento e subsequentemente recidivaram após o pós-tratamento acompanhamento). Os indivíduos que foram negativos na Semana 12 foram tratados por 48 semanas e acompanhados por 24 semanas após o tratamento. A resposta ao tratamento foi definida como HCV-RNA indetectável 24 semanas após o tratamento (medido usando um teste baseado em pesquisa, limite de detecção de 125 UI/mL). A taxa de resposta geral foi de 22% (497/2293) (IC 99%: 19,5, 23,9). Indivíduos com as seguintes características eram menos propensos a se beneficiar do retratamento: não resposta anterior, tratamento anterior com interferon peguilado, fibrose ou cirrose em ponte significativa e infecção pelo genótipo 1.
As taxas de resposta virológica sustentada de retratamento por características basais estão resumidas na Tabela 16.
A obtenção de um HCV-RNA indetectável na semana 12 do tratamento foi um forte preditor de RVS. Neste estudo, 1.470 (64%) indivíduos não atingiram um HCV-RNA indetectável na semana 12 do tratamento e foram oferecidas inscrições em estudos de tratamento de longo prazo, devido a uma resposta inadequada ao tratamento. Dos 823 (36%) indivíduos que eram HCV-RNA indetectáveis na Semana 12 de tratamento, aqueles infectados com o genótipo 1 tiveram uma SVR de 48% (245/507), com uma gama de respostas por pontuações de fibrose (F4-F2) de 39-55%. Indivíduos infectados com genótipo 2/3 que eram HCV-RNA indetectáveis na semana 12 do tratamento tiveram uma RVS geral de 70% (196/281), com uma faixa de respostas por pontuações de fibrose (F4-F2) de 60-83%. Para todos os genótipos, pontuações mais altas de fibrose foram associadas a uma menor probabilidade de atingir SVR.
Sujeitos Pediátricos
Indivíduos pediátricos não tratados anteriormente de 3 a 17 anos de idade com hepatite C crônica compensada e HCV-RNA detectável foram tratados com REBETOL 15 mg/kg por dia e PegIntron 60 mcg/m² uma vez por semana por 24 ou 48 semanas com base no genótipo do HCV e na linha de base viral carregar. Todos os indivíduos deveriam ser acompanhados por 24 semanas pós-tratamento. Um total de 107 indivíduos receberam tratamento, dos quais 52% eram do sexo feminino, 89% eram caucasianos e 67% estavam infectados com HCV Genótipo 1. Indivíduos infectados com Genótipos 1, 4 ou Genótipo 3 com HCV-RNA maior ou igual a 600.000 UI/mL receberam 48 semanas de terapia enquanto aqueles infectados com Genótipo 2 ou Genótipo 3 com HCV-RNA menor que 600.000 UI/mL receberam 24 semanas de terapia. Os resultados do ensaio estão resumidos na Tabela 17.
Terapia combinada REBETOL/INTRON A
Assuntos adultos
Assuntos não tratados anteriormente
Adultos com hepatite C crônica compensada e HCV-RNA detectável (avaliados por um laboratório central usando um ensaio de RT-PCR baseado em pesquisa) que não foram tratados anteriormente com terapia com interferon alfa foram inscritos em dois estudos multicêntricos, duplo-cegos (EUA e internacional) e randomizados para receber REBETOL 200 mg cápsulas 1.200 mg/dia (1.000 mg/dia para indivíduos com peso menor ou igual a 75 kg) e INTRON A 3 MIU três vezes por semana ou INTRON A e placebo por 24 ou 48 semanas seguido por 24 semanas de acompanhamento fora da terapia. O estudo internacional não continha um braço de tratamento com INTRON A de 24 semanas e placebo. O estudo norte-americano envolveu 912 indivíduos que, no início do estudo, eram 67% do sexo masculino, 89% caucasianos com uma pontuação média de Knodell HAI (I+II+III) de 7,5 e 72% do genótipo 1. O estudo internacional, realizado na Europa, Israel , Canadá e Austrália, inscreveram 799 indivíduos (65% do sexo masculino, 95% caucasianos, pontuação média de Knodell 6,8 e 58% genótipo 1).
Os resultados do ensaio estão resumidos na Tabela 18.
Dos indivíduos que não atingiram HCV-RNA abaixo do limite de detecção do ensaio baseado em pesquisa na Semana 24 do tratamento com REBETOL/INTRON A, menos de 5% responderam a mais 24 semanas de tratamento combinado.
Entre os indivíduos com HCV Genótipo 1 tratados com terapia REBETOL/INTRON A que alcançaram HCV-RNA abaixo do limite de detecção do ensaio baseado em pesquisa por 24 semanas, aqueles randomizados para 48 semanas de tratamento tiveram respostas virológicas mais altas em comparação com aqueles no grupo de 24-24 semanas. grupo de tratamento semana. Não houve aumento observado nas taxas de resposta para indivíduos com HCV não genótipo 1 randomizados para terapia com REBETOL/INTRON A por 48 semanas em comparação com 24 semanas.
Assuntos de recaída
Indivíduos com hepatite C crônica compensada e HCV-RNA detectável (avaliado por um laboratório central usando um ensaio de RT-PCR baseado em pesquisa) que recaíram após um ou dois ciclos de terapia com interferon (definido como níveis séricos anormais de ALT) foram inscritos em dois multicêntrico, duplo-cego (EUA e internacional) e randomizado para receber REBETOL 1.200 mg/dia (1.000 mg/dia para indivíduos com peso ≤ 75 kg) e INTRON A 3 MIU três vezes por semana ou INTRON A e placebo por 24 semanas, seguido de 24 semanas de acompanhamento sem terapia. O estudo norte-americano envolveu 153 indivíduos que, no início do estudo, eram 67% do sexo masculino, 92% caucasianos com uma pontuação média de Knodell HAI (I+II+III) de 6,8 e 58% do genótipo 1. O estudo internacional, realizado na Europa, Israel , Canadá e Austrália, inscreveram 192 indivíduos (64% do sexo masculino, 95% caucasianos, pontuação média de Knodell 6,6 e 56% genótipo 1). Os resultados do ensaio estão resumidos na Tabela 19.
As respostas virológicas e histológicas foram semelhantes entre indivíduos do sexo masculino e feminino em ambos os ensaios previamente não tratados e de recidiva.
Sujeitos Pediátricos
Indivíduos pediátricos de 3 a 16 anos de idade com hepatite C crônica compensada e HCV-RNA detectável (avaliados por um laboratório central usando um ensaio de RT-PCR baseado em pesquisa) foram tratados com REBETOL 15 mg/kg por dia e INTRON A 3 MUI/ m² três vezes por semana durante 48 semanas seguidas de 24 semanas de acompanhamento sem terapia. Um total de 118 indivíduos recebeu tratamento, dos quais 57% eram homens, 80% caucasianos e 78% genótipo 1. Indivíduos com menos de 5 anos de idade receberam REBETOL solução oral e aqueles com 5 anos de idade ou mais receberam REBETOL solução oral ou cápsulas.
Os resultados do ensaio estão resumidos na Tabela 20.
Indivíduos com genótipo viral 1, independentemente da carga viral, tiveram uma taxa de resposta mais baixa à terapia combinada INTRON A/REBETOL em comparação com indivíduos com genótipo não-1, 36% vs. 81%. Indivíduos com ambos os fatores de mau prognóstico (genótipo 1 e alta carga viral) tiveram uma taxa de resposta de 26% (13/50).
INFORMAÇÃO DO PACIENTE
Nenhuma informação fornecida. Por favor, consulte o AVISOS E PRECAUÇÕES seção.