Xeloda 500mg Capecitabine Uso, efeitos colaterais e dosagem. Preço na farmácia online. Medicamentos genericos sem receita.
O que é Xeloda 500mg e como é usado?
Xeloda é um medicamento de prescrição usado para tratar os sintomas de câncer, como câncer de cólon, câncer colorretal e câncer de mama. Xeloda pode ser usado sozinho ou com outros medicamentos.
Xeloda pertence a uma classe de medicamentos chamados Antineoplásicos, Antimetabólitos.
Não se sabe se Xeloda 500mg é seguro e eficaz em crianças.
Quais são os possíveis efeitos colaterais do Xeloda 500mg?
Xeloda pode causar efeitos colaterais graves, incluindo:
- febre acima de 100,5 graus,
- náusea,
- perda de apetite,
- comer muito menos do que o habitual,
- vômitos (mais de uma vez em 24 horas),
- diarreia grave (mais de 4 vezes por dia ou durante a noite),
- bolhas ou úlceras na boca,
- gengivas vermelhas ou inchadas,
- dificuldade para engolir,
- dor, sensibilidade, vermelhidão, inchaço, bolhas ou descamação da pele nas mãos ou pés,
- sentindo muita sede ou calor,
- não conseguir urinar,
- suor intenso,
- pele quente e seca,
- dor ou pressão no peito,
- batimentos cardíacos irregulares,
- falta de ar,
- inchaço ou ganho de peso rápido,
- micção dolorosa ou difícil,
- inchaço nos pés ou tornozelos,
- sentindo-se cansado,
- falta de ar,
- urina escura,
- fezes cor de barro,
- amarelecimento da pele ou dos olhos (icterícia),
- febre ou outros sintomas de gripe,
- tosse,
- feridas na pele,
- pele pálida,
- contusões fáceis,
- sangramento incomum,
- sentindo-se tonto,
- batimentos cardíacos rápidos,
- dor de garganta,
- inchaço no rosto ou na língua,
- queimando em seus olhos, e
- dor na pele que é seguida por uma erupção cutânea vermelha ou roxa (especialmente na face ou na parte superior do corpo) e causa bolhas e descamação
Obtenha ajuda médica imediatamente, se tiver algum dos sintomas listados acima.
Os efeitos colaterais mais comuns de Xeloda 500mg incluem:
- dor de estômago,
- constipação,
- dor de estômago,
- sensação de cansaço,
- erupção cutânea leve e
- dormência ou formigamento nas mãos ou pés
Informe o médico se tiver algum efeito colateral que o incomode ou que não desapareça.
Estes não são todos os possíveis efeitos colaterais do Xeloda. Para mais informações, consulte seu médico ou farmacêutico.
Ligue para o seu médico para aconselhamento médico sobre os efeitos colaterais. Você pode relatar efeitos colaterais ao FDA em 1-800-FDA-1088.
AVISO
INTERAÇÃO XELODA-WARFARINA
XELODA 500mg Interação de Varfarina: Pacientes recebendo concomitantemente capecitabina e terapia anticoagulante derivada de cumarina oral devem ter sua resposta anticoagulante (INR ou tempo de protrombina) monitorada frequentemente para ajustar a dose de anticoagulante adequadamente. Uma interação medicamentosa XELODA-Warfarina clinicamente importante foi demonstrada em um estudo de farmacologia clínica [ver AVISOS E PRECAUÇÕES INTERAÇÕES DE DROGAS]. Parâmetros de coagulação alterados e/ou sangramento, incluindo morte, foram relatados em pacientes tomando XELODA 500mg concomitantemente com anticoagulantes derivados de cumarina, como varfarina e fenprocumona. Os relatórios pós-comercialização mostraram aumentos clinicamente significativos no tempo de protrombina (TP) e INR em pacientes que foram estabilizados com anticoagulantes no momento em que XELODA 500mg foi introduzido. Esses eventos ocorreram dentro de vários dias e até vários meses após o início da terapia com XELODA 500mg e, em alguns casos, dentro de 1 mês após a interrupção do XELODA. Esses eventos ocorreram em pacientes com e sem metástases hepáticas. A idade superior a 60 anos e o diagnóstico de câncer predispõem independentemente os pacientes a um risco aumentado de coagulopatia.
DESCRIÇÃO
XELODA (capecitabina) é um carbamato de fluoropirimidina com atividade antineoplásica. É uma pró-droga sistêmica administrada por via oral de 5'-desoxi-5-fluorouridina (5'-DFUR) que é convertida em 5-fluorouracil.
nome químico da capecitabina é 5'-desoxi-5-fluoro-N-[(pentiloxi) carbonil]-citidina e tem um peso molecular de 359,35. Capecitabina tem a seguinte fórmula estrutural:
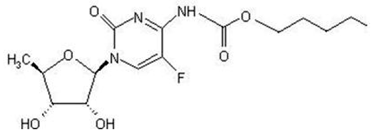
Capecitabina é um pó cristalino branco a esbranquiçado com uma solubilidade aquosa de 26 mg/mL a 20°C.
XELODA é fornecido na forma de comprimidos revestidos por película oblongos e biconvexos para administração oral. Cada comprimido cor de pêssego claro contém 150 mg de capecitabina e cada comprimido cor de pêssego contém 500 mg de capecitabina. Os ingredientes inativos de XELODA incluem: lactose anidra, croscarmelose sódica, hidroxipropilmetilcelulose, celulose microcristalina, estearato de magnésio e água purificada. O revestimento de película de pêssego ou pêssego claro contém hidroxipropilmetilcelulose, talco, dióxido de titânio e óxidos de ferro sintéticos amarelos e vermelhos.
INDICAÇÕES
Câncer colorretal
- XELODA é indicado como agente único para tratamento adjuvante em pacientes com câncer de cólon Dukes' C que foram submetidos à ressecção completa do tumor primário quando o tratamento apenas com fluoropirimidina é preferido. XELODA 500mg não foi inferior a 5fluorouracil e leucovorina (5-FU/LV) para sobrevida livre de doença (DFS). Os médicos devem considerar os resultados de ensaios de quimioterapia combinada, que mostraram melhora na DFS e OS, ao prescrever o agente único XELODA 500mg no tratamento adjuvante do câncer de cólon Dukes' C.
- XELODA 500mg é indicado como tratamento de primeira linha de pacientes com carcinoma colorretal metastático quando o tratamento apenas com fluoropirimidina é preferido. A quimioterapia de combinação mostrou um benefício de sobrevivência em comparação com 5-FU/LV sozinho. Um benefício de sobrevida em relação ao 5-FU/LV não foi demonstrado com a monoterapia com XELODA 500mg. O uso de XELODA em vez de 5FU/LV em combinações não foi adequadamente estudado para garantir a segurança ou preservação da vantagem de sobrevivência.
Câncer de mama
- XELODA em combinação com docetaxel é indicado para o tratamento de pacientes com câncer de mama metastático após falha de quimioterapia prévia contendo antraciclina.
- XELODA 500 mg em monoterapia também é indicado para o tratamento de pacientes com câncer de mama metastático resistente tanto ao paclitaxel quanto a um regime de quimioterapia contendo antraciclina ou resistente ao paclitaxel e para os quais a terapia adicional com antraciclina não é indicada (por exemplo, pacientes que receberam doses cumulativas de 400 mg/m2 de doxorrubicina ou equivalentes de doxorrubicina). A resistência é definida como doença progressiva durante o tratamento, com ou sem resposta inicial, ou recidiva dentro de 6 meses após a conclusão do tratamento com um regime adjuvante contendo antraciclina.
DOSAGEM E ADMINISTRAÇÃO
Instruções importantes de administração
Os comprimidos de XELODA devem ser engolidos inteiros com água dentro de 30 minutos após uma refeição. XELODA 500mg é um medicamento citotóxico. Siga os procedimentos especiais aplicáveis de manuseio e descarte.1 Se os comprimidos de XELODA 500mg precisarem ser cortados ou esmagados, isso deve ser feito por um profissional treinado no manuseio seguro de medicamentos citotóxicos, usando equipamentos e procedimentos de segurança apropriados. A dose de XELODA é calculada de acordo com a área de superfície corporal.
Dose Inicial Padrão
Monoterapia (Câncer Colorretal Metastático, Câncer Colorretal Adjuvante, Câncer de Mama Metastático)
A dose recomendada de XELODA é de 1.250 mg/m2 administrado por via oral duas vezes ao dia (manhã e noite; equivalente a 2.500 mg/m2 dose diária total) por 2 semanas, seguido por um período de descanso de 1 semana administrado em ciclos de 3 semanas (ver ).
tratamento adjuvante em pacientes com câncer de cólon Dukes' C é recomendado por um total de 6 meses [ou seja, XELODA 1250 mg/m2 por via oral duas vezes ao dia por 2 semanas seguido por um período de descanso de 1 semana, administrado em ciclos de 3 semanas para um total de 8 ciclos (24 semanas)].
Em combinação com docetaxel (câncer de mama metastático)
Em combinação com docetaxel, a dose recomendada de XELODA é de 1.250 mg/m2 duas vezes ao dia por 2 semanas, seguido por um período de repouso de 1 semana, combinado com docetaxel a 75 mg/m2 em infusão intravenosa de 1 hora a cada 3 semanas. A pré-medicação, de acordo com a bula do docetaxel, deve ser iniciada antes da administração do docetaxel para pacientes recebendo a combinação de XELODA mais docetaxel. exibe a dose diária total de XELODA por área de superfície corporal e o número de comprimidos a serem tomados em cada dose.
Diretrizes de Gerenciamento de Dose
Em geral
dosagem de XELODA pode precisar ser individualizada para otimizar o manejo do paciente. Os pacientes devem ser cuidadosamente monitorados quanto à toxicidade e as doses de XELODA 500mg devem ser modificadas conforme necessário para acomodar a tolerância individual do paciente ao tratamento. Estudos clínicos ]. A toxicidade devido à administração de XELODA 500mg pode ser controlada por tratamento sintomático, interrupção da dose e ajuste da dose de XELODA. Uma vez que a dose tenha sido reduzida, ela não deve ser aumentada posteriormente. As doses de XELODA 500mg omitidas por toxicidade não são substituídas ou restauradas; em vez disso, o paciente deve retomar os ciclos de tratamento planejados.
A dose de fenitoína e a dose de anticoagulantes derivados de cumarina podem precisar ser reduzidas quando qualquer um dos medicamentos é administrado concomitantemente com XELODA [ver INTERAÇÕES MEDICAMENTOSAS ].
Monoterapia (Câncer Colorretal Metastático, Câncer Colorretal Adjuvante, Câncer de Mama Metastático)
O esquema de modificação de dose de XELODA conforme descrito abaixo (ver ) é recomendado para o manejo de reações adversas.
Em combinação com docetaxel (câncer de mama metastático)
As modificações de dose de XELODA 500mg para toxicidade devem ser feitas de acordo com o acima para XELODA. No início de um ciclo de tratamento, se for indicado um atraso no tratamento para XELODA 500 mg ou docetaxel, a administração de ambos os agentes deve ser adiada até que os requisitos para reiniciar os dois medicamentos sejam atendidos.
O esquema de redução da dose de docetaxel quando usado em combinação com XELODA para o tratamento de câncer de mama metastático é mostrado na Tabela 3.
Ajuste da dose inicial em populações especiais
Insuficiência renal
Nenhum ajuste na dose inicial de XELODA 500mg é recomendado em pacientes com insuficiência renal leve (clearance de creatinina = 51 a 80 mL/min [Cockroft e Gault, como mostrado abaixo]). Em pacientes com insuficiência renal moderada (depuração de creatinina basal = 30 a 50 mL/min), uma redução da dose para 75% da dose inicial de XELODA quando usado como monoterapia ou em combinação com docetaxel (de 1250 mg/m2 a 950 mg/m2 duas vezes ao dia) é recomendado [ver Uso em populações específicas e FARMACOLOGIA CLÍNICA ]. O ajuste subsequente da dose é recomendado conforme descrito em e (dependendo do regime) se um paciente desenvolver um evento adverso de grau 2 a 4 [ver AVISOS E PRECAUÇÕES ]. As recomendações de ajuste de dose inicial para pacientes com insuficiência renal moderada aplicam-se tanto a XELODA 500 mg em monoterapia quanto a XELODA 500 mg em uso combinado com docetaxel.
Equação de Cockroft e Gault:
Geriatria
Os médicos devem ter cautela ao monitorar os efeitos de XELODA 500mg em idosos. Dados insuficientes estão disponíveis para fornecer uma recomendação de dosagem.
COMO FORNECIDO
Formas de Dosagem Forças
XELODA é fornecido na forma de comprimidos revestidos por película oblongos e biconvexos para administração oral. Cada comprimido cor de pêssego claro contém 150 mg de capecitabina e cada comprimido cor de pêssego contém 500 mg de capecitabina.
150 mg
Cor: pêssego claro Gravação: XELODA de um lado e 150 do outro comprimidos de 150 mg são embalados em frascos de 60 ( NDC 0004-1100-20), embalados individualmente em caixa de papelão.
500 mg
Cor: Pêssego Gravação: XELODA 500mg de um lado e 500 do outro 500mg comprimidos são embalados em frascos de 120 ( NDC 0004-1101-50), embalados individualmente em caixa de papelão.
Armazenamento e manuseio
Armazenar a 25°C (77°F); excursões permitidas de 15° a 30°C (59° a 86°F). [Consulte a temperatura ambiente controlada da USP]. MANTENHA BEM FECHADO.
XELODA é uma droga citotóxica. Siga os procedimentos especiais aplicáveis de manuseio e descarte.1 Qualquer produto não utilizado deve ser descartado de acordo com os requisitos locais ou programas de devolução de medicamentos.
REFERÊNCIAS
1. “Medicamentos Perigosos da OSHA.” OSHA. http://www.osha.gov/SLTC/hazardousdrugs/index.html.
Distribuído por: Genentech USA, Inc. Membro do Grupo Roche, 1 DNA Way, South San Francisco, CA 94080-4990. Revisado: maio de 2021
EFEITOS COLATERAIS
Como os ensaios clínicos são conduzidos em condições muito variadas, as taxas de reações adversas observadas nos ensaios clínicos de um medicamento não podem ser diretamente comparadas às taxas nos ensaios clínicos de outro medicamento e podem não refletir as taxas observadas na prática.
Câncer de cólon adjuvante
mostra as reações adversas que ocorrem em ≥5% dos pacientes de um estudo de fase 3 em pacientes com câncer de cólon Dukes' C que receberam pelo menos uma dose da medicação do estudo e tiveram pelo menos uma avaliação de segurança. Um total de 995 pacientes foi tratado com 1.250 mg/m2 duas vezes ao dia de XELODA 500 mg administrado por 2 semanas seguido por um período de descanso de 1 semana, e 974 pacientes receberam 5-FU e leucovorina (20 mg/m2 leucovorina IV seguido de 425 mg/m2 IV em bolus 5FU nos dias 1-5 a cada 28 dias). A duração média do tratamento foi de 164 dias para pacientes tratados com capecitabina e 145 dias para pacientes tratados com 5-FU/LV. Um total de 112 (11%) e 73 (7%) pacientes tratados com capecitabina e 5-FU/LV, respectivamente, descontinuaram o tratamento devido a reações adversas. Um total de 18 mortes por todas as causas ocorreram no estudo ou dentro de 28 dias após o recebimento do medicamento em estudo: 8 (0,8%) pacientes randomizados para XELODA 500mg e 10 (1,0%) randomizados para 5-FU/LV.
mostra anormalidades laboratoriais de grau 3/4 ocorrendo em ≥1% dos pacientes de um estudo de fase 3 em pacientes com câncer de cólon Dukes' C que receberam pelo menos uma dose da medicação do estudo e tiveram pelo menos uma avaliação de segurança.
Câncer Colorretal Metastático
Monoterapia
mostra as reações adversas que ocorrem em ≥5% dos pacientes do agrupamento dos dois estudos de fase 3 em câncer colorretal metastático de primeira linha. Um total de 596 pacientes com câncer colorretal metastático foi tratado com 1250 mg/m2 duas vezes ao dia de XELODA administrado por 2 semanas seguido por um período de descanso de 1 semana, e 593 pacientes receberam 5-FU e leucovorina no regime Mayo (20 mg/m2 de leucovorina IV seguida de 425 mg/m2 de bolus IV de 5-FU, nos dias 1-5, a cada 28 dias). No banco de dados colorretal agrupado, a duração média do tratamento foi de 139 dias para pacientes tratados com capecitabina e 140 dias para pacientes tratados com 5-FU/LV. Um total de 78 (13%) e 63 (11%) pacientes tratados com capecitabina e 5-FU/LV, respectivamente, descontinuaram o tratamento devido a reações adversas/doença intercorrente. Um total de 82 mortes por todas as causas ocorreram no estudo ou dentro de 28 dias após o recebimento do medicamento em estudo: 50 (8,4%) pacientes randomizados para XELODA 500mg e 32 (5,4%) randomizados para 5-FU/LV.
Câncer de mama
Em combinação com docetaxel
Os dados a seguir são mostrados para o estudo de combinação com XELODA 500mg e docetaxel em pacientes com câncer de mama metastático em e . No braço da combinação de XELODA e docetaxel, o tratamento foi XELODA administrado por via oral 1.250 mg/m2 duas vezes ao dia como terapia intermitente (2 semanas de tratamento seguidas de 1 semana sem tratamento) por pelo menos 6 semanas e docetaxel administrado como infusão intravenosa de 1 hora em uma dose de 75 mg/m2 no primeiro dia de cada ciclo de 3 semanas durante pelo menos 6 semanas. No braço de monoterapia, o docetaxel foi administrado como uma infusão intravenosa de 1 hora na dose de 100 mg/m2 no primeiro dia de cada ciclo de 3 semanas por pelo menos 6 semanas. A duração média do tratamento foi de 129 dias no braço de combinação e 98 dias no braço de monoterapia. Um total de 66 pacientes (26%) no braço de combinação e 49 (19%) no braço de monoterapia se retiraram do estudo devido a reações adversas. A porcentagem de pacientes que necessitaram de reduções de dose devido a reações adversas foi de 65% no braço de combinação e 36% no braço de monoterapia. A percentagem de doentes que necessitaram de interrupções do tratamento devido a reações adversas no braço de combinação foi de 79%. As interrupções do tratamento faziam parte do esquema de modificação de dose para o braço de terapia combinada, mas não para os pacientes tratados com docetaxel em monoterapia.
Monoterapia
Os dados a seguir são mostrados para o estudo em pacientes com câncer de mama em estágio IV que receberam uma dose de 1250 mg/m2 administrada duas vezes ao dia por 2 semanas seguidas por um período de descanso de 1 semana. A duração média do tratamento foi de 114 dias. Um total de 13 dos 162 pacientes (8%) descontinuaram o tratamento devido a reações adversas/doença intercorrente.
Eventos Adversos Clinicamente Relevantes em
Os eventos adversos clinicamente relevantes relatados em
Monoterapia (Câncer Colorretal Metastático, Câncer Colorretal Adjuvante, Câncer de Mama Metastático)
Gastrointestinal: distensão abdominal, disfagia, proctalgia, ascite (0,1%), úlcera gástrica (0,1%), íleo (0,3%), dilatação tóxica do intestino, gastroenterite (0,1%)
Pele e Subcutâneo.: distúrbio ungueal (0,1%), sudorese aumentada (0,1%), reação de fotossensibilidade (0,1%), ulceração da pele, prurido, síndrome de recordação de radiação (0,2%)
Em geral: dor no peito (0,2%), doença semelhante à gripe, afrontamentos, dor (0,1%), rouquidão, irritabilidade, dificuldade em andar, sede, massa torácica, colapso, fibrose (0,1%), hemorragia, edema, sedação
Neurológico: insônia, ataxia (0,5%), tremor, disfasia, encefalopatia (0,1%), coordenação anormal, disartria, perda de consciência (0,2%), equilíbrio prejudicado
Metabolismo: aumento de peso, caquexia (0,4%), hipertrigliceridemia (0,1%), hipocalemia, hipomagnesemia
Olho: conjuntivite
Respiratório: tosse (0,1%), epistaxe (0,1%), asma (0,2%), hemoptise, desconforto respiratório (0,1%), dispneia
Cardíaco: taquicardia (0,1%), bradicardia, fibrilação atrial, extrassístoles ventriculares, extrassístoles, miocardite (0,1%), derrame pericárdico
Infecções: laringite (1,0%), bronquite (0,2%), pneumonia (0,2%), broncopneumonia (0,2%), ceratoconjuntivite, sepse (0,3%), infecções fúngicas (incluindo candidíase) (0,2%)
Musculoesquelético: mialgia, dor óssea (0,1%), artrite (0,1%), fraqueza muscular
Sangue e Linfático: leucopenia (0,2%), distúrbio de coagulação (0,1%), depressão da medula óssea (0,1%), púrpura trombocitopenia idiopática (1,0%), pancitopenia (0,1%)
Vascular: hipotensão (0,2%), hipertensão (0,1%), linfedema (0,1%), embolia pulmonar (0,2%), acidente vascular cerebral (0,1%)
Psiquiátrico: depressão, confusão (0,1%)
Renal: insuficiência renal (0,6%)
Orelha: vertigem
Hepatobiliar: fibrose hepática (0,1%), hepatite (0,1%), hepatite colestática (0,1%), testes de função hepática anormais
Sistema imunológico: hipersensibilidade a drogas (0,1%)
XELODA 500mg em combinação com docetaxel (câncer de mama metastático)
Gastrointestinal: íleo (0,4%), enterocolite necrosante (0,4%), úlcera esofágica (0,4%), diarreia hemorrágica (0,8%)
Neurológico: ataxia (0,4%), síncope (1,2%), perda do paladar (0,8%), polineuropatia (0,4%), enxaqueca (0,4%)
Cardíaco: taquicardia supraventricular (0,4%)
Infecção: sepse neutropênica (2,4%), sepse (0,4%), broncopneumonia (0,4%) Sangue e
Linfático: agranulocitose (0,4%), diminuição da protrombina (0,4%)
Vascular: hipotensão (1,2%), flebite venosa e tromboflebite (0,4%), hipotensão postural (0,8%)
Renal: insuficiência renal (0,4%)
Hepatobiliar: icterícia (0,4%), testes de função hepática anormais (0,4%), insuficiência hepática (0,4%), coma hepático (0,4%), hepatotoxicidade (0,4%)
Sistema imunológico: hipersensibilidade (1,2%)
Experiência pós-marketing
As seguintes reações adversas foram observadas no cenário pós-comercialização: angioedema, insuficiência hepática, estenose do ducto lacrimal, insuficiência renal aguda secundária à desidratação, incluindo desfecho fatal. AVISOS E PRECAUÇÕES ], lúpus eritematoso cutâneo, distúrbios da córnea, incluindo ceratite, leucoencefalopatia tóxica, reações cutâneas graves, como síndrome de Stevens-Johnson e necrólise epidérmica tóxica (NET) [ver AVISOS E PRECAUÇÕES ], a síndrome mão-e-pé persistente ou grave pode eventualmente levar à perda de impressões digitais [ver AVISOS E PRECAUÇÕES ]
Em casos de exposição a comprimidos triturados de XELODA 500mg, foram relatadas as seguintes reações adversas: irritação e inchaço nos olhos, erupção cutânea, diarreia, parestesia, dor de cabeça, irritação gástrica, vômito e náusea.
INTERAÇÕES MEDICAMENTOSAS
Interações Medicamentosas
Anticoagulantes
Parâmetros de coagulação alterados e/ou sangramento foram relatados em pacientes tomando XELODA 500mg concomitantemente com anticoagulantes derivados de cumarina, como varfarina e fenprocumon [ver AVISO DE CAIXA ]. Esses eventos ocorreram dentro de vários dias e até vários meses após o início da terapia com XELODA 500mg e, em alguns casos, dentro de 1 mês após a interrupção do XELODA. Esses eventos ocorreram em pacientes com e sem metástases hepáticas. Em um estudo de interação medicamentosa com administração de dose única de varfarina, houve um aumento significativo na AUC média da S-varfarina [ver FARMACOLOGIA CLÍNICA ]. O valor máximo de INR observado aumentou 91%. Esta interação deve-se provavelmente à inibição do citocromo P450 2C9 pela capecitabina e/ou seus metabólitos.
Fenitoína
O nível de fenitoína deve ser cuidadosamente monitorado em pacientes tomando XELODA 500mg e a dose de fenitoína pode precisar ser reduzida [ver DOSAGEM E ADMINISTRAÇÃO ]. Relatórios pós-comercialização indicam que alguns pacientes que receberam XELODA 500mg e fenitoína apresentaram toxicidade associada a níveis elevados de fenitoína. Não foram realizados estudos formais de interação medicamentosa com fenitoína, mas presume-se que o mecanismo de interação seja a inibição da isoenzima CYP2C9 pela capecitabina e/ou seus metabólitos.
Leucovorina
A concentração de 5-fluorouracil é aumentada e sua toxicidade pode ser aumentada pela leucovorina. Mortes por enterocolite grave, diarreia e desidratação foram relatadas em pacientes idosos recebendo semanalmente leucovorina e fluorouracil.
Substratos CYP2C9
Além da varfarina, não foram realizados estudos formais de interação medicamentosa entre XELODA e outros substratos do CYP2C9. Deve-se ter cuidado quando XELODA é coadministrado com substratos CYP2C9.
Alopurinol
O uso concomitante com alopurinol pode diminuir a concentração dos metabólitos ativos da capecitabina [ver FARMACOLOGIA CLÍNICA ], o que pode diminuir a eficácia de XELODA. Evite o uso de alopurinol durante o tratamento com XELODA.
Interação medicamento-alimento
Foi demonstrado que os alimentos reduzem tanto a taxa quanto a extensão da absorção da capecitabina [ver FARMACOLOGIA CLÍNICA ]. Em todos os ensaios clínicos, os pacientes foram instruídos a administrar XELODA 500mg dentro de 30 minutos após uma refeição. Recomenda-se que XELODA seja administrado com alimentos [ver DOSAGEM E ADMINISTRAÇÃO ].
AVISOS
Incluído como parte do "PRECAUÇÕES" Seção
PRECAUÇÕES
Coagulopatia
Pacientes recebendo concomitantemente capecitabina e terapia anticoagulante com derivado de cumarina oral devem ter sua resposta anticoagulante (INR ou tempo de protrombina) monitorada de perto com grande frequência e a dose de anticoagulante deve ser ajustada de acordo. AVISO DE CAIXA e INTERAÇÕES MEDICAMENTOSAS ].
Diarréia
XELODA 500mg pode induzir diarréia, às vezes grave. Pacientes com diarreia grave devem ser cuidadosamente monitorados e receber reposição de fluidos e eletrólitos se ficarem desidratados. Em 875 pacientes com câncer de mama metastático ou colorretal que receberam monoterapia com XELODA, o tempo médio para a primeira ocorrência de diarreia de grau 2 a 4 foi de 34 dias (variação de 1 a 369 dias). A duração média da diarreia de grau 3 a 4 foi de 5 dias. A diarreia grau 2 do National Cancer Institute of Canada (NCIC) é definida como um aumento de 4 a 6 evacuações/dia ou evacuações noturnas, diarreia grau 3 como um aumento de 7 a 9 evacuações/dia ou incontinência e má absorção e diarreia grau 4 como um aumento de ≥10 evacuações/dia ou diarreia com sangue grosseiro ou a necessidade de suporte parenteral. Se ocorrer diarreia de grau 2, 3 ou 4, a administração de XELODA deve ser imediatamente interrompida até que a diarreia desapareça ou diminua de intensidade para grau 1 [ver DOSAGEM E ADMINISTRAÇÃO ]. Tratamentos antidiarreicos padrão (por exemplo, loperamida) são recomendados.
Enterocolite necrosante (tiflite) foi relatada.
Cardiotoxicidade
A cardiotoxicidade observada com XELODA 500mg inclui infarto/isquemia do miocárdio, angina, arritmias, parada cardíaca, insuficiência cardíaca, morte súbita, alterações eletrocardiográficas e cardiomiopatia. Essas reações adversas podem ser mais comuns em pacientes com história prévia de doença arterial coronariana.
Deficiência de Dihidropirimidina Desidrogenase
Com base em relatórios pós-comercialização, pacientes com certas mutações homozigotas ou heterozigotas compostas no gene DPD que resultam na ausência completa ou quase completa da atividade da DPD têm risco aumentado de toxicidade aguda precoce e efeitos adversos graves, com risco de vida ou fatais. reações causadas por XELODA (por exemplo, mucosite, diarreia, neutropenia e neurotoxicidade). Pacientes com atividade parcial de DPD também podem ter risco aumentado de reações adversas graves, fatais ou com risco de vida causadas por XELODA.
Suspenda ou descontinue permanentemente XELODA 500 mg com base na avaliação clínica do início, duração e gravidade das toxicidades observadas em pacientes com evidência de início precoce agudo ou toxicidade incomumente grave, que pode indicar ausência quase completa ou total de atividade de DPD. Nenhuma dose de XELODA provou ser segura para pacientes com ausência completa de atividade de DPD. Não há dados suficientes para recomendar uma dose específica em pacientes com atividade parcial de DPD medida por qualquer teste específico.
Desidratação e Insuficiência Renal
Desidratação foi observada e pode causar insuficiência renal aguda que pode ser fatal. Pacientes com função renal comprometida preexistente ou que estejam recebendo XELODA concomitantemente com agentes nefrotóxicos conhecidos estão em maior risco. Os doentes com anorexia, astenia, náuseas, vómitos ou diarreia podem ficar rapidamente desidratados. Monitore os pacientes quando XELODA 500mg é administrado para prevenir e corrigir a desidratação no início. Se ocorrer desidratação de grau 2 (ou superior), o tratamento com XELODA deve ser imediatamente interrompido e a desidratação corrigida. O tratamento não deve ser reiniciado até que o paciente seja reidratado e quaisquer causas precipitantes tenham sido corrigidas ou controladas. Modificações de dose devem ser aplicadas para o evento adverso precipitante conforme necessário [ver DOSAGEM E ADMINISTRAÇÃO ].
Pacientes com insuficiência renal moderada no início do estudo requerem redução da dose [ver DOSAGEM E ADMINISTRAÇÃO ]. Pacientes com insuficiência renal leve e moderada na linha de base devem ser cuidadosamente monitorados quanto a reações adversas. A interrupção imediata da terapia com ajustes de dose subsequentes é recomendada se um paciente desenvolver um evento adverso de grau 2 a 4, conforme descrito em [ver DOSAGEM E ADMINISTRAÇÃO , Uso em populações específicas , e FARMACOLOGIA CLÍNICA ].
Toxicidade Embrio-Fetal
Com base em resultados de estudos de reprodução em animais e seu mecanismo de ação, XELODA pode causar danos ao feto quando administrado a uma mulher grávida [ver FARMACOLOGIA CLÍNICA ]. Os dados limitados disponíveis não são suficientes para informar o uso de XELODA 500mg em mulheres grávidas. Em estudos de reprodução animal, a administração de capecitabina a animais prenhes durante o período de organogênese causou embrioletalidade e teratogenicidade em camundongos e embrioletalidade em macacos em 0,2 e 0,6 vezes a exposição (AUC) em pacientes que receberam a dose recomendada, respectivamente [ver Uso em populações específicas ]. Informar as mulheres grávidas sobre o risco potencial para o feto. Aconselhar as mulheres com potencial reprodutivo a usar contracepção eficaz durante o tratamento e por 6 meses após a última dose de XELODA [ver Uso em populações específicas ].
Toxicidade Mucocutânea e Dermatológica
Reações mucocutâneas graves, algumas com desfecho fatal, como síndrome de Stevens-Johnson e necrólise epidérmica tóxica (NET) podem ocorrer em pacientes tratados com XELODA [ver REAÇÕES ADVERSAS ]. XELODA 500mg deve ser descontinuado permanentemente em pacientes que apresentarem uma reação mucocutânea grave possivelmente atribuível ao tratamento com XELODA.
síndrome mão-pé (eritrodisestesia palmo-plantar ou eritema acral induzido por quimioterapia) é uma toxicidade cutânea. O tempo médio para início foi de 79 dias (variando de 11 a 360 dias) com uma faixa de gravidade de graus 1 a 3 para pacientes que receberam monoterapia com XELODA 500mg no cenário metastático. O grau 1 é caracterizado por qualquer um dos seguintes: dormência, disestesia/parestesia, formigamento, inchaço indolor ou eritema das mãos e/ou pés e/ou desconforto que não interrompa as atividades normais. A síndrome mão-pé de grau 2 é definida como eritema doloroso e inchaço das mãos e/ou pés e/ou desconforto que afeta as atividades de vida diária do paciente. A síndrome mão-pé de grau 3 é definida como descamação úmida, ulceração, bolhas ou dor intensa nas mãos e/ou pés e/ou desconforto grave que faz com que o paciente seja incapaz de trabalhar ou realizar atividades da vida diária. Síndrome mão-pé persistente ou grave (grau 2 e acima) pode eventualmente levar à perda de impressões digitais que podem afetar a identificação do paciente. Se ocorrer síndrome mão-pé de grau 2 ou 3, a administração de XELODA deve ser interrompida até que o evento resolva ou diminua em intensidade para grau 1. Após síndrome mão-pé grau 3, as doses subsequentes de XELODA 500mg devem ser reduzidas [ Vejo DOSAGEM E ADMINISTRAÇÃO ].
Hiperbilirrubinemia
Em 875 pacientes com câncer de mama metastático ou colorretal que receberam pelo menos uma dose de XELODA 1.250 mg/m2 duas vezes ao dia como monoterapia por 2 semanas seguidas por um período de repouso de 1 semana, ocorreu hiperbilirrubinemia grau 3 (1,5-3 x LSN) em 15,2% (n=133) dos pacientes e hiperbilirrubinemia grau 4 (>3 x LSN) ocorreu em 3,9% (n=34) dos pacientes. Dos 566 pacientes que apresentavam metástases hepáticas no início do estudo e 309 pacientes sem metástases hepáticas no início do estudo, hiperbilirrubinemia grau 3 ou 4 ocorreu em 22,8% e 12,3%, respectivamente. Dos 167 pacientes com hiperbilirrubinemia grau 3 ou 4, 18,6% (n=31) também tiveram elevações pós-basais (graus 1 a 4, sem elevações na linha de base) na fosfatase alcalina e 27,5% (n=46) tiveram elevações pós-basais nas transaminases em qualquer momento (não necessariamente simultâneo). A maioria desses pacientes, 64,5% (n=20) e 71,7% (n=33), apresentava metástases hepáticas no início do estudo. Além disso, 57,5% (n = 96) e 35,3% (n = 59) dos 167 pacientes apresentaram elevações (graus 1 a 4) tanto na linha de base quanto na linha de base na fosfatase alcalina ou transaminases, respectivamente. Apenas 7,8% (n=13) e 3,0% (n=5) apresentaram elevações de grau 3 ou 4 na fosfatase alcalina ou transaminases.
Nos 596 pacientes tratados com XELODA 500mg como terapia de primeira linha para câncer colorretal metastático, a incidência de hiperbilirrubinemia grau 3 ou 4 foi semelhante ao banco de dados geral de segurança de ensaios clínicos da monoterapia com XELODA 500mg. O tempo médio para o início da hiperbilirrubinemia de grau 3 ou 4 na população com câncer colorretal foi de 64 dias e a mediana da bilirrubina total aumentou de 8 μm/L na linha de base para 13 μm/L durante o tratamento com XELODA. Dos 136 pacientes com câncer colorretal com hiperbilirrubinemia grau 3 ou 4, 49 pacientes tinham hiperbilirrubinemia grau 3 ou 4 como seu último valor medido, dos quais 46 tinham metástases hepáticas no início do estudo.
Em 251 pacientes com câncer de mama metastático que receberam uma combinação de XELODA 500mg e docetaxel, ocorreu hiperbilirrubinemia grau 3 (1,5 a 3 x LSN) em 7% (n=17) e hiperbilirrubinemia grau 4 (>3 x LSN) em 2% (n=5).
Se ocorrerem elevações da bilirrubina de grau 3 a 4 relacionadas ao medicamento, a administração de XELODA 500 mg deve ser imediatamente interrompida até que a hiperbilirrubinemia diminua para ≤ 3,0 X LSN [ver modificações de dose recomendadas em DOSAGEM E ADMINISTRAÇÃO ].
Hematologico
Em 875 pacientes com câncer de mama metastático ou câncer colorretal que receberam uma dose de 1.250 mg/m2 administrada duas vezes ao dia como monoterapia por 2 semanas seguidas por um período de repouso de 1 semana, 3,2%, 1,7% e 2,4% dos pacientes apresentaram grau 3 ou 4 neutropenia, trombocitopenia ou diminuição da hemoglobina, respectivamente. Em 251 pacientes com câncer de mama metastático que receberam uma dose de XELODA em combinação com docetaxel, 68% tiveram neutropenia grau 3 ou 4, 2,8% tiveram trombocitopenia grau 3 ou 4 e 9,6% tiveram anemia grau 3 ou 4.
Pacientes com contagem basal de neutrófilos
Pacientes Geriátricos
Pacientes ≥80 anos de idade podem apresentar uma maior incidência de reações adversas de grau 3 ou 4. Em 875 pacientes com câncer de mama ou colorretal metastático que receberam XELODA 500mg em monoterapia, 62% dos 21 pacientes ≥80 anos de idade tratados com XELODA 500mg apresentaram um evento adverso grau 3 ou 4 relacionado ao tratamento: diarreia em 6 (28,6%) , náuseas em 3 (14,3%), síndrome mão-pé em 3 (14,3%) e vômitos em 2 (9,5%) pacientes. Entre os 10 pacientes com 70 anos de idade ou mais (nenhum paciente tinha >80 anos de idade) tratados com XELODA 500mg em combinação com docetaxel, 30% (3 em 10) dos pacientes apresentaram diarreia e estomatite de grau 3 ou 4 e 40 % (4 em 10) experimentou síndrome mão-pé grau 3.
Entre os 67 pacientes ≥60 anos de idade recebendo XELODA em combinação com docetaxel, a incidência de reações adversas de grau 3 ou 4 relacionadas ao tratamento, reações adversas graves relacionadas ao tratamento, descontinuações devido a reações adversas, descontinuações do tratamento devido a reações adversas e tratamento descontinuações nos dois primeiros ciclos de tratamento foi maior do que no grupo de pacientes
Em 995 pacientes recebendo XELODA como terapia adjuvante para câncer de cólon Dukes C após ressecção do tumor primário, 41% dos 398 pacientes ≥65 anos de idade tratados com XELODA experimentaram um evento adverso grau 3 ou 4 relacionado ao tratamento: -síndrome do pé em 75 (18,8%), diarreia em 52 (13,1%), estomatite em 12 (3,0%), neutropenia/granulocitopenia em 11 (2,8%), vômito em 6 (1,5%) e náusea em 5 (1,3%) %) pacientes. Em pacientes ≥65 anos de idade (toda a população randomizada; capecitabina 188 pacientes, 5-FU/LV 208 pacientes) tratados para câncer de cólon Dukes' C após ressecção do tumor primário, as taxas de risco para sobrevida livre de doença e sobrevida global para XELODA em comparação com 5-FU/LV foram 1,01 (IC 95% 0,80 – 1,27) e 1,04 (IC 95% 0,79 – 1,37), respectivamente.
Insuficiência Hepática
Pacientes com disfunção hepática leve a moderada devido a metástases hepáticas devem ser cuidadosamente monitorados quando XELODA for administrado. O efeito da disfunção hepática grave na eliminação de XELODA não é conhecido [ver Uso em populações específicas e FARMACOLOGIA CLÍNICA ].
Combinação com outras drogas
O uso de XELODA em combinação com irinotecano não foi adequadamente estudado.
Informações de Aconselhamento do Paciente
Aconselhe o paciente a ler o rótulo do paciente aprovado pela FDA ( INFORMAÇÃO DO PACIENTE ).
Diarréia
Informe os pacientes com diarreia de grau 2 (um aumento de 4 a 6 evacuações/dia ou evacuações noturnas) ou mais ou com diarreia sanguinolenta grave com dor abdominal intensa e febre para parar de tomar XELODA. Aconselhar os pacientes sobre o uso de tratamentos antidiarreicos (por exemplo, loperamida) para controlar a diarreia [ver AVISOS E PRECAUÇÕES ].
Cardiotoxicidade
Aconselhar os pacientes sobre o risco de cardiotoxicidade e contatar imediatamente seu médico ou ir a um pronto-socorro para um novo início de dor no peito, falta de ar, tontura ou vertigens [ver AVISOS E PRECAUÇÕES ].
Deficiência de Dihidropirimidina Desidrogenase
Aconselhe os pacientes a notificar seu médico se tiverem uma deficiência conhecida de DPD. Aconselhar os pacientes se eles tiverem ausência completa ou quase completa de atividade de DPD, eles estão em risco aumentado de toxicidade aguda precoce e reações adversas graves, com risco de vida ou fatais causadas por XELODA (por exemplo, mucosite, diarreia, neutropenia e neurotoxicidade) [ver AVISOS E PRECAUÇÕES ].
Desidratação e Insuficiência Renal
Instrua os pacientes com desidratação de grau 2 ou superior (indicação de fluidos intravenosos AVISOS E PRECAUÇÕES ].
Instruções importantes de administração
Aconselhe os pacientes a engolir os comprimidos de XELODA 500mg inteiros com água dentro de 30 minutos após uma refeição. Aconselhe os pacientes e cuidadores a não esmagar ou cortar os comprimidos de XELODA. Aconselhe os pacientes se não conseguirem engolir os comprimidos de XELODA inteiros, para informar seu médico [ver DOSAGEM E ADMINISTRAÇÃO ].
Hipersensibilidade e angioedema
Avise os pacientes que XELODA pode causar reações de hipersensibilidade graves e angioedema. Aconselhar os pacientes com hipersensibilidade conhecida à capecitabina ou 5-fluorouracil a informar seu médico [ver CONTRA-INDICAÇÕES ]. Instruir os pacientes que desenvolvem reações de hipersensibilidade ou sintomas mucocutâneos (por exemplo, urticária, erupção cutânea, eritema, prurido ou inchaço da face, lábios, língua ou garganta que dificultam a deglutição ou respiração) a parar de tomar XELODA e entrar em contato imediatamente com seu médico ou ir ao pronto-socorro. [Vejo REAÇÕES ADVERSAS ].
Náusea
Instruir os pacientes com náusea de grau 2 (ingestão de alimentos significativamente diminuída, mas capaz de comer de forma intermitente) ou superior a parar de tomar XELODA 500mg imediatamente e entrar em contato com seu médico para tratamento da náusea [ver REAÇÕES ADVERSAS ].

Vômito
Instruir os pacientes que apresentarem vômitos de grau 2 (2 a 5 episódios em um período de 24 horas) ou mais a parar de tomar XELODA imediatamente e entrar em contato com seu médico para tratamento do vômito [ver REAÇÕES ADVERSAS ].
Síndrome mão e pé
Instruir os pacientes com síndrome mão-pé grau 2 (eritema doloroso e inchaço das mãos e/ou pés e/ou desconforto que afeta as atividades da vida diária do paciente) ou superior a parar de tomar XELODA 500mg imediatamente e entrar em contato com seu médico . Informe os pacientes que o início do tratamento sintomático é recomendado, e a síndrome mão-pé pode levar à perda de impressões digitais que podem afetar a identificação pessoal [ver REAÇÕES ADVERSAS ].
Estomatite
Informe os pacientes com estomatite grau 2 (eritema doloroso, edema ou úlceras da boca ou língua, mas capaz de comer) ou superior para parar de tomar XELODA imediatamente e entrar em contato com seu médico [ver REAÇÕES ADVERSAS ].
Febre e Neutropenia
Informe os pacientes que desenvolverem febre de 100,5°F ou mais ou outra evidência de infecção potencial para entrar em contato com seu médico [ver REAÇÕES ADVERSAS ].
Toxicidade Embrio-Fetal
Aconselhar as mulheres com potencial reprodutivo sobre o risco potencial para o feto e usar métodos contraceptivos eficazes durante o tratamento com XELODA 500mg e por 6 meses após a última dose. Aconselhe as mulheres a informar seu médico sobre uma gravidez conhecida ou suspeita [ver AVISOS E PRECAUÇÕES , Uso em populações específicas ].
Aconselhar os pacientes do sexo masculino com parceiras femininas com potencial reprodutivo a usarem métodos contraceptivos eficazes durante o tratamento com XELODA 500mg e por 3 meses após a última dose [ver Uso em populações específicas ].
Lactação
Aconselhe as mulheres a não amamentar durante o tratamento com XELODA 500mg e por 2 semanas após a última dose [ver Uso em populações específicas ].
Toxicologia não clínica
Carcinogênese, Mutagênese, Prejuízo da Fertilidade
Não foram realizados estudos adequados investigando o potencial carcinogênico da capecitabina. Capecitabina não foi mutagênica in vitro para bactérias (teste de Ames) ou células de mamíferos (ensaio de mutação genética V79/HPRT de hamster chinês). Capecitabina foi clastogênica in vitro para linfócitos de sangue periférico humano, mas não clastogênica in vivo para medula óssea de camundongo (teste de micronúcleo). Fluorouracil causa mutações em bactérias e leveduras. Fluorouracil também causa anormalidades cromossômicas no teste de micronúcleo de camundongo in vivo.
Em estudos de fertilidade e desempenho reprodutivo geral em camundongos fêmeas, doses orais de capecitabina de 760 mg/kg/dia (cerca de 2300 mg/m2/dia) perturbaram o estro e, consequentemente, causaram uma diminuição na fertilidade. Em camundongos que engravidaram, nenhum feto sobreviveu a essa dose. A perturbação no estro foi reversível. Nos machos, esta dose causou alterações degenerativas nos testículos, incluindo diminuição do número de espermatócitos e espermátides. Em estudos farmacocinéticos separados, esta dose em camundongos produziu valores de AUC de 5'-DFUR cerca de 0,7 vezes os valores correspondentes em pacientes que receberam a dose diária recomendada.
Uso em populações específicas
Gravidez
Resumo do risco
Com base em achados em estudos de reprodução animal e seu mecanismo de ação, XELODA 500mg pode causar dano fetal quando administrado a uma mulher grávida [ver FARMACOLOGIA CLÍNICA ]. Os dados humanos limitados disponíveis não são suficientes para informar o risco associado ao medicamento durante a gravidez. Em estudos de reprodução animal, a administração de capecitabina a animais prenhes durante o período de organogênese causou letalidade embrionária e teratogenicidade em camundongos e letalidade embrionária em macacos em 0,2 e 0,6 vezes a exposição (AUC) em pacientes que receberam a dose recomendada, respectivamente [ver Dados ]. Informar as mulheres grávidas sobre o risco potencial para o feto.
O risco de fundo estimado de defeitos congênitos graves e aborto espontâneo para a população indicada é desconhecido. Na população geral dos EUA, o risco de fundo estimado de defeitos congênitos graves e aborto espontâneo em gestações clinicamente reconhecidas é de 2-4% e 15-20%, respectivamente.
Dados
Dados de animais
administração oral de capecitabina a camundongos prenhes durante o período de organogênese na dose de 198 mg/kg/dia causou malformações e letalidade embrionária. Em estudos farmacocinéticos separados, esta dose em camundongos produziu valores de AUC de 5'-DFUR que foram aproximadamente 0,2 vezes os valores de AUC em pacientes que receberam a dose diária recomendada. As malformações em camundongos incluíram fenda palatina, anoftalmia, microftalmia, oligodactilia, polidactilia, sindactilia, cauda kinky e dilatação dos ventrículos cerebrais. A administração oral de capecitabina a macacas grávidas durante o período de organogênese na dose de 90 mg/kg/dia, causou letalidade fetal. Esta dose produziu valores de AUC de 5'-DFUR que foram aproximadamente 0,6 vezes os valores de AUC em pacientes que receberam a dose diária recomendada.
Lactação
Resumo do risco
Não há informações sobre a presença de capecitabina no leite humano, ou sobre seus efeitos na produção de leite ou no lactente. Os metabólitos da capecitabina estavam presentes no leite de camundongos lactantes [ver Dados ]. Devido ao potencial de reações adversas graves da exposição à capecitabina em bebês amamentados, aconselhe as mulheres a não amamentar durante o tratamento com XELODA 500 mg e por 2 semanas após a dose final.
Dados
Camundongos lactantes que receberam uma dose oral única de capecitabina excretaram quantidades significativas de metabólitos de capecitabina no leite.
Fêmeas e Machos com Potencial Reprodutivo
Teste de gravidez
O teste de gravidez é recomendado para mulheres com potencial reprodutivo antes de iniciar XELODA.
Contracepção
Mulheres
XELODA 500mg pode causar dano fetal quando administrado a uma mulher grávida [ver Gravidez ]. Aconselhe as mulheres com potencial reprodutivo a usar contracepção eficaz durante o tratamento e por 6 meses após a dose final de XELODA.
Machos
Com base nos achados de toxicidade genética, aconselhe os pacientes do sexo masculino com parceiras do sexo feminino com potencial reprodutivo a usar contracepção eficaz durante o tratamento e por 3 meses após a dose final de XELODA [ver Toxicologia não clínica ].
Infertilidade
Com base em estudos em animais, XELODA pode prejudicar a fertilidade em fêmeas e machos com potencial reprodutivo [ver Toxicologia não clínica ].
Uso Pediátrico
segurança e eficácia de XELODA em pacientes pediátricos não foram estabelecidas. Nenhum benefício clínico foi demonstrado em dois ensaios de braço único em pacientes pediátricos com gliomas de tronco cerebral recém-diagnosticados e gliomas de alto grau. Em ambos os estudos, os pacientes pediátricos receberam uma formulação pediátrica experimental de capecitabina concomitantemente e após a conclusão da radioterapia (dose total de 5580 cGy em frações de 180 cGy). A biodisponibilidade relativa da formulação experimental para XELODA 500mg foi semelhante.
primeiro estudo foi realizado em 22 pacientes pediátricos (idade média de 8 anos, intervalo de 5-17 anos) com gliomas de tronco cerebral difusos intrínsecos não disseminados recém-diagnosticados e gliomas de alto grau. Na parte de determinação da dose do estudo, os pacientes receberam capecitabina com radioterapia concomitante em doses variando de 500 mg/m2 a 850 mg/m2 a cada 12 horas por até 9 semanas. Após um intervalo de 2 semanas, os pacientes receberam 1250 mg/m2 de capecitabina a cada 12 horas nos dias 1-14 de um ciclo de 21 dias por até 3 ciclos. A dose máxima tolerada (DMT) de capecitabina administrada concomitantemente com radioterapia foi de 650 mg/m2 a cada 12 horas. As principais toxicidades limitantes da dose foram eritrodisestesia palmo-plantar e elevação da alanina aminotransferase (ALT).
segundo estudo foi realizado em 34 pacientes pediátricos adicionais com gliomas intrínsecos difusos de tronco encefálico não disseminados recém-diagnosticados (idade média de 7 anos, intervalo de 3 a 16 anos) e 10 pacientes pediátricos que receberam o MTD de capecitabina no estudo de determinação de dose e atingiram os critérios de elegibilidade para este ensaio. Todos os pacientes receberam 650 mg/m2 de capecitabina a cada 12 horas com radioterapia concomitante por até 9 semanas. Após um intervalo de 2 semanas, os pacientes receberam 1250 mg/m2 de capecitabina a cada 12 horas nos dias 1-14 de um ciclo de 21 dias por até 3 ciclos.
Não houve melhora na taxa de sobrevida livre de progressão em um ano e na taxa de sobrevida global em um ano em pacientes pediátricos com gliomas intrínsecos de tronco encefálico recém-diagnosticados que receberam capecitabina em relação a uma população semelhante de pacientes pediátricos que participaram de outros ensaios clínicos.
perfil de reações adversas da capecitabina foi consistente com o perfil de reações adversas conhecidas em adultos, com exceção de anormalidades laboratoriais que ocorreram mais comumente em pacientes pediátricos. As anormalidades laboratoriais mais frequentemente relatadas (incidência por paciente ≥40%) foram ALT aumentada (75%), linfocitopenia (73%), leucopenia (73%), hipocalemia (68%), trombocitopenia (57%), hipoalbuminemia (55%) %), neutropenia (50%), hematócrito baixo (50%), hipocalcemia (48%), hipofosfatemia (45%) e hiponatremia (45%).
Uso Geriátrico
Os médicos devem prestar atenção especial ao monitoramento dos efeitos adversos de XELODA 500mg em idosos [ver AVISOS E PRECAUÇÕES ].
Insuficiência Hepática
Tenha cuidado quando pacientes com disfunção hepática leve a moderada devido a metástases hepáticas são tratados com XELODA. O efeito da disfunção hepática grave em XELODA não é conhecido [ver AVISOS E PRECAUÇÕES e FARMACOLOGIA CLÍNICA ].
Insuficiência renal
Pacientes com insuficiência renal moderada (depuração de creatinina = 30 a 50 mL/min) e grave (depuração de creatinina CONTRA-INDICAÇÕES , AVISOS E PRECAUÇÕES , DOSAGEM E ADMINISTRAÇÃO , e FARMACOLOGIA CLÍNICA ].
SOBREDOSAGEM
As manifestações de sobredosagem aguda incluem náuseas, vómitos, diarreia, irritação e hemorragia gastrointestinal e depressão da medula óssea. A gestão médica da sobredosagem deve incluir intervenções médicas de suporte habituais destinadas a corrigir as manifestações clínicas apresentadas. Embora não tenha sido relatada nenhuma experiência clínica usando diálise como tratamento para superdosagem de XELODA 500 mg, a diálise pode ser benéfica na redução das concentrações circulantes de 5'-DFUR, um metabólito de baixo peso molecular do composto original.
Doses únicas de XELODA 500 mg não foram letais para camundongos, ratos e macacos em doses de até 2.000 mg/kg (2,4, 4,8 e 9,6 vezes a dose diária humana recomendada com base em mg/m2).
CONTRA-INDICAÇÕES
Insuficiência Renal Grave
XELODA é contraindicado em pacientes com insuficiência renal grave (clearance de creatinina abaixo de 30 mL/min [Cockroft e Gault]) [ver Uso em populações específicas e FARMACOLOGIA CLÍNICA ].
Hipersensibilidade
XELODA 500mg é contraindicado em pacientes com hipersensibilidade conhecida à capecitabina ou a qualquer um de seus componentes. XELODA é contraindicado em pacientes com hipersensibilidade conhecida ao 5fluorouracil.
FARMACOLOGIA CLÍNICA
Mecanismo de ação
As enzimas convertem a capecitabina em 5-fluorouracil (5-FU) in vivo. Tanto as células normais como as tumorais metabolizam 5-FU em monofosfato de 5-fluoro-2'-desoxiuridina (FdUMP) e trifosfato de 5-fluorouridina (FUTP). Esses metabólitos causam lesão celular por dois mecanismos diferentes. Primeiro, FdUMP e o cofator folato, N5-10-metilenotetrahidrofolato, ligam-se à timidilato sintase (TS) para formar um complexo ternário ligado covalentemente. Esta ligação inibe a formação de timidilato a partir de 2'-desoxiuridilato. O timidilato é o precursor necessário do trifosfato de timidina, que é essencial para a síntese do DNA, de modo que uma deficiência deste composto pode inibir a divisão celular. Em segundo lugar, as enzimas transcricionais nucleares podem incorporar erroneamente FUTP no lugar do trifosfato de uridina (UTP) durante a síntese de RNA. Esse erro metabólico pode interferir no processamento do RNA e na síntese de proteínas.
Farmacocinética
Absorção
Após a administração oral de 1.255 mg/m2 BID a pacientes com câncer, a capecitabina atingiu os níveis sanguíneos máximos em cerca de 1,5 horas (Tmax) com os níveis máximos de 5-FU ocorrendo um pouco mais tarde, em 2 horas. Os alimentos reduziram a taxa e a extensão da absorção da capecitabina com Cmax e AUC0-∞ médias diminuídas em 60% e 35%, respectivamente. A Cmax e AUC0-∞ de 5-FU também foram reduzidas por alimentos em 43% e 21%, respectivamente. A comida atrasou o Tmax de ambos os pais e 5-FU em 1,5 horas [ver AVISOS E PRECAUÇÕES , DOSAGEM E ADMINISTRAÇÃO , e Interação medicamento-alimento ].
A farmacocinética de XELODA 500mg e seus metabólitos foram avaliados em cerca de 200 pacientes com câncer em uma faixa de dosagem de 500 a 3.500 mg/m2/dia. Acima desta faixa, a farmacocinética de XELODA e seu metabólito, 5'-DFCR foram proporcionais à dose e não mudaram ao longo do tempo. Os aumentos nas AUCs de 5'-DFUR e 5-FU, no entanto, foram maiores do que proporcionais ao aumento da dose e a AUC de 5-FU foi 34% maior no dia 14 do que no dia 1. A variabilidade interpaciente no A Cmax e a AUC de 5-FU foram superiores a 85%.
Distribuição
ligação às proteínas plasmáticas da capecitabina e seus metabólitos é inferior a 60% e não é dependente da concentração. Capecitabina foi principalmente ligada à albumina humana (aproximadamente 35%). XELODA 500mg tem um baixo potencial para interações farmacocinéticas relacionadas à ligação às proteínas plasmáticas.
Bioativação e Metabolismo
capecitabina é extensivamente metabolizada enzimaticamente em 5-FU. No fígado, uma carboxilesterase de 60 kDa hidrolisa grande parte do composto em 5'-desoxi-5-fluorocitidina (5'-DFCR). A citidina desaminase, uma enzima encontrada na maioria dos tecidos, incluindo tumores, converte subsequentemente 5'-DFCR em 5'-DFUR. A enzima, timidina fosforilase (dThdPase), então hidrolisa 5'DFUR para a droga ativa 5-FU. Muitos tecidos em todo o corpo expressam timidina fosforilase. Alguns carcinomas humanos expressam esta enzima em concentrações mais elevadas do que os tecidos normais circundantes. Após a administração oral de XELODA 7 dias antes da cirurgia em pacientes com câncer colorretal, a razão mediana da concentração de 5-FU em tumores colorretais para tecidos adjacentes foi de 2,9 (variação de 0,9 a 8,0). Essas proporções não foram avaliadas em pacientes com câncer de mama ou comparadas à infusão de 5-FU.
Via metabólica da capecitabina para 5-FU
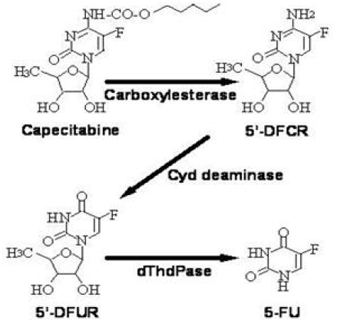
enzima dihidropirimidina desidrogenase hidrogena 5-FU, o produto do metabolismo da capecitabina, para o muito menos tóxico 5-fluoro-5,6-dihidro-fluorouracil (FUH2). A dihidropirimidinase cliva o anel de pirimidina para produzir ácido 5-fluoro-ureido-propiônico (FUPA). Finalmente, a β-ureido-propionase cliva FUPA em α-fluoro-β-alanina (FBAL), que é eliminada na urina.
Estudos enzimáticos in vitro com microssomas hepáticos humanos indicaram que a capecitabina e seus metabólitos (5'-DFUR, 5'-DFCR, 5-FU e FBAL) não inibiram o metabolismo dos substratos de teste pelas isoenzimas 1A2, 2A6, 3A4, citocromo P450, 2C19, 2D6 e 2E1.
Excreção
Capecitabina e seus metabólitos são predominantemente excretados na urina; 95,5% da dose administrada de capecitabina é recuperada na urina. A excreção fecal é mínima (2,6%). O principal metabólito excretado na urina é o FBAL que representa 57% da dose administrada. Cerca de 3% da dose administrada é excretada na urina como droga inalterada. A meia-vida de eliminação da capecitabina original e do 5-FU foi de cerca de 0,75 horas.
Efeito da idade, sexo e raça na farmacocinética da Capecitabina
Uma análise populacional de dados agrupados de dois grandes estudos controlados em pacientes com câncer colorretal metastático (n=505) que receberam XELODA 500mg a 1.250 mg/m2 duas vezes ao dia indicou que sexo (202 mulheres e 303 homens) e raça (455 pacientes brancos/caucasianos, 22 pacientes negros e 28 pacientes de outra raça) não têm influência na farmacocinética de 5'DFUR, 5-FU e FBAL. A idade não tem influência significativa na farmacocinética de 5'-DFUR e 5-FU na faixa de 27 a 86 anos. Um aumento de 20% na idade resulta em um aumento de 15% na AUC de FBAL [ver AVISOS E PRECAUÇÕES e DOSAGEM E ADMINISTRAÇÃO ].
Após a administração oral de 825 mg/m2 de capecitabina duas vezes ao dia por 14 dias, os pacientes japoneses (n=18) apresentaram Cmax cerca de 36% menor e AUC 24% menor para capecitabina do que os pacientes caucasianos (n=22). Os pacientes japoneses também apresentaram Cmax cerca de 25% menor e AUC 34% menor para FBAL do que os pacientes caucasianos. O significado clínico dessas diferenças é desconhecido. Não ocorreram diferenças significativas na exposição a outros metabólitos (5'-DFCR, 5'-DFUR e 5FU).
Efeito da insuficiência hepática
XELODA foi avaliado em 13 pacientes com disfunção hepática leve a moderada devido a metástases hepáticas definidas por um escore composto incluindo bilirrubina, AST/ALT e fosfatase alcalina após uma dose única de 1255 mg/m2 de XELODA. Tanto a AUC0-∞ como a Cmax da capecitabina aumentaram 60% em doentes com disfunção hepática em comparação com doentes com função hepática normal (n=14). A AUC0-∞ e Cmax de 5-FU não foram afetadas. Em pacientes com disfunção hepática leve a moderada devido a metástases hepáticas, deve-se ter cautela ao administrar XELODA 500mg. O efeito da disfunção hepática grave em XELODA 500mg não é conhecido [ver AVISOS E PRECAUÇÕES e Uso em populações especiais ].
Efeito da insuficiência renal
Após administração oral de 1.250 mg/m2 de capecitabina duas vezes ao dia a pacientes com câncer com vários graus de insuficiência renal, pacientes com insuficiência renal moderada (depuração de creatinina = 30 a 50 mL/min) e grave (depuração de creatinina 80 mL/min). A exposição sistêmica ao 5'-DFUR foi 42% e 71% maior em pacientes com insuficiência renal moderada e grave, respectivamente, do que em pacientes normais. A exposição sistêmica à capecitabina foi cerca de 25% maior em pacientes com insuficiência renal moderada e grave. DOSAGEM E ADMINISTRAÇÃO , CONTRA-INDICAÇÕES , AVISOS E PRECAUÇÕES , e Uso em populações especiais ].
Efeito da Capecitabina na Farmacocinética da Varfarina
Em quatro pacientes com câncer, a administração crônica de capecitabina (1.250 mg/m2 bid) com uma dose única de 20 mg de varfarina aumentou a AUC média da S-varfarina em 57% e diminuiu sua depuração em 37%. A AUC de INR corrigida na linha de base nesses 4 pacientes aumentou 2,8 vezes, e o valor de INR médio máximo observado foi aumentado em 91% [ver AVISO DE CAIXA e INTERAÇÕES MEDICAMENTOSAS ].
Efeito dos Antiácidos na Farmacocinética da Capecitabina
Quando Maalox® (20 mL), um antiácido contendo hidróxido de alumínio e hidróxido de magnésio, foi administrado imediatamente após XELODA (1250 mg/m2, n=12 pacientes com câncer), a AUC e a Cmax aumentaram 16% e 35%, respectivamente, para capecitabina e em 18% e 22%, respectivamente, para 5'-DFCR. Nenhum efeito foi observado nos outros três principais metabólitos (5'-DFUR, 5-FU, FBAL) de XELODA.
Efeito do alopurinol na Capecitabina
literatura publicada relatou que o uso concomitante com alopurinol pode diminuir a conversão da capecitabina nos metabólitos ativos, FdUMP e FUTP; no entanto, o significado clínico não foi totalmente caracterizado.
Efeito da Capecitabina na farmacocinética do docetaxel e vice-versa
Um estudo de Fase 1 avaliou o efeito do XELODA na farmacocinética do docetaxel (Taxotere®) e o efeito do docetaxel na farmacocinética do XELODA foi realizado em 26 pacientes com tumores sólidos. Verificou-se que XELODA não tem efeito sobre a farmacocinética do docetaxel (Cmax e AUC) e o docetaxel não tem efeito sobre a farmacocinética da capecitabina e do precursor 5-FU 5'-DFUR.
Estudos clínicos
Câncer de cólon adjuvante
Um ensaio clínico multicêntrico randomizado e controlado de fase 3 em pacientes com câncer de cólon Dukes C (X-ACT) forneceu dados sobre o uso de XELODA para o tratamento adjuvante de pacientes com câncer de cólon. O objetivo primário do estudo foi comparar a sobrevida livre de doença (DFS) em pacientes que receberam XELODA com aqueles que receberam apenas 5-FU/LV IV. Neste estudo, 1.987 pacientes foram randomizados para tratamento com XELODA 1.250 mg/m2 por via oral duas vezes ao dia por 2 semanas seguido de um período de descanso de 1 semana, administrado em ciclos de 3 semanas para um total de 8 ciclos (24 semanas) ou IV bolus 5-FU 425 mg/m2 e 20 mg/m2 de leucovorina IV nos dias 1 a 5, administrados em ciclos de 4 semanas para um total de 6 ciclos (24 semanas). Os pacientes do estudo deveriam ter entre 18 e 75 anos de idade com câncer de cólon em estágio C de Dukes confirmado histologicamente com pelo menos um linfonodo positivo e ter sido submetido (dentro de 8 semanas antes da randomização) ressecção completa do tumor primário sem evidência macroscópica ou microscópica de tumor remanescente. Os pacientes também foram obrigados a não ter quimioterapia ou imunoterapia citotóxica prévia (exceto esteróides) e ter um status de desempenho ECOG de 0 ou 1 (KPS ≥ 70%), ANC ≥ 1,5x109/L, plaquetas ≥ 100x109/L, creatinina sérica ≤ 1,5 LSN, bilirrubina total ≤ 1,5 LSN, AST/ALT ≤ 2,5 LSN e CEA dentro dos limites normais no momento da randomização.
Os dados demográficos da linha de base para pacientes com XELODA e 5-FU/LV são mostrados na Tabela 10. As características da linha de base foram bem equilibradas entre os braços.
Todos os pacientes com função renal normal ou insuficiência renal leve iniciaram o tratamento com a dose inicial completa de 1250 mg/m2 por via oral duas vezes ao dia. A dose inicial foi reduzida em pacientes com insuficiência renal moderada (depuração de creatinina calculada de 30 a 50 mL/min) no início do estudo [ver DOSAGEM E ADMINISTRAÇÃO ]. Posteriormente, para todos os pacientes, as doses foram ajustadas quando necessário de acordo com a toxicidade. A gestão da dose de XELODA 500mg incluiu reduções de dose, atrasos no ciclo e interrupções do tratamento (ver Tabela 11).
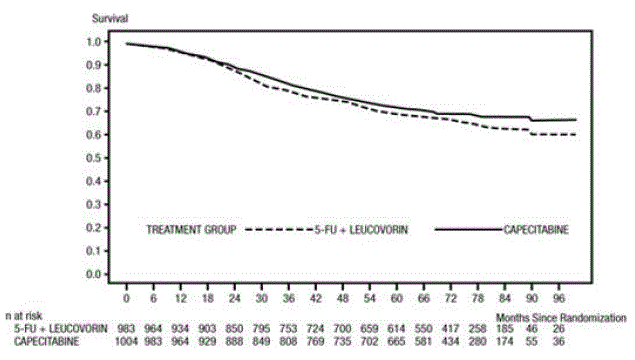
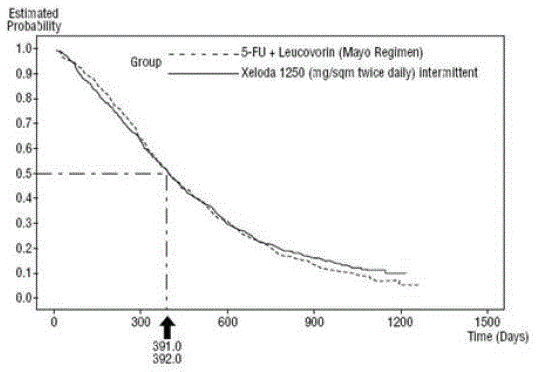
A mediana de seguimento no momento da análise foi de 83 meses (6,9 anos). A taxa de risco para DFS para XELODA 500mg em comparação com 5-FU/LV foi de 0,88 (IC 95% 0,77 - 1,01) (ver e figura 1 ). Como o limite superior de confiança de 95% de 95% da taxa de risco foi inferior a 1,20, o XELODA não foi inferior ao 5-FU/LV. A escolha da margem de não inferioridade de 1,20 corresponde à retenção de aproximadamente 75% do efeito 5-FU/LV no DFS. A taxa de risco para XELODA em comparação com 5-FU/LV em relação à sobrevida global foi de 0,86 (IC 95% 0,74 – 1,01). As taxas de sobrevida global em 5 anos foram de 71,4% para XELODA e 68,4% para 5-FU/LV (ver Figura 2).
Figura 1 Estimativas Kaplan-Meier de Sobrevivência Livre de Doenças (Toda a População Randomizada)a
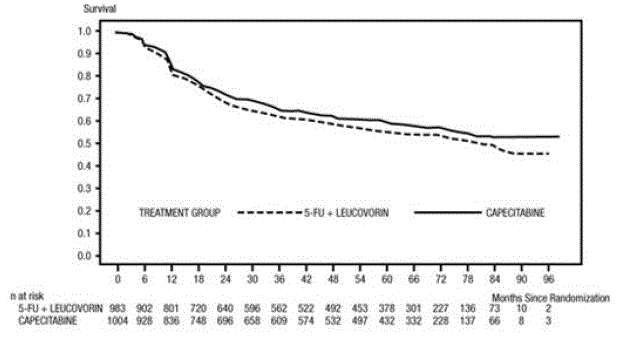
Figura 2 Estimativas Kaplan-Meier de Sobrevivência Geral (Toda a População Randomizada)
Câncer Colorretal Metastático
Em geral
dose recomendada de XELODA 500mg foi determinada em um estudo clínico randomizado, aberto, explorando a eficácia e segurança da terapia contínua com capecitabina (1331 mg/m2/dia em duas doses divididas, n=39), terapia intermitente com capecitabina ( 2510 mg/m2/dia em duas doses divididas, n=34), e terapia intermitente com capecitabina em combinação com leucovorina oral (LV) (capecitabina 1657 mg/m2/dia em duas doses divididas, n=35; leucovorina 60 mg/ dia) em pacientes com carcinoma colorretal avançado e/ou metastático no cenário metastático de primeira linha. Não houve vantagem aparente na taxa de resposta ao adicionar leucovorina ao XELODA; no entanto, a toxicidade foi aumentada. XELODA, 1250 mg/m2 duas vezes ao dia por 14 dias, seguido de 1 semana de descanso, foi selecionado para desenvolvimento clínico adicional com base no perfil geral de segurança e eficácia dos três esquemas estudados.
Monoterapia
Dados de dois ensaios clínicos abertos, multicêntricos, randomizados e controlados envolvendo 1.207 pacientes apoiam o uso de XELODA no tratamento de primeira linha de pacientes com carcinoma colorretal metastático. Os dois estudos clínicos foram idênticos em design e foram conduzidos em 120 centros em diferentes países. O Estudo 1 foi realizado nos EUA, Canadá, México e Brasil; O Estudo 2 foi realizado na Europa, Israel, Austrália, Nova Zelândia e Taiwan. Ao todo, em ambos os ensaios, 603 pacientes foram randomizados para tratamento com XELODA 500mg na dose de 1.250 mg/m2 duas vezes ao dia por 2 semanas seguido por um período de descanso de 1 semana e administrado em ciclos de 3 semanas; 604 pacientes foram randomizados para tratamento com 5-FU e leucovorina (20 mg/m2 de leucovorina IV seguido de 425 mg/m2 de bolus IV de 5-FU, nos dias 1 a 5, a cada 28 dias).
Em ambos os ensaios, a sobrevida global, o tempo de progressão e a taxa de resposta (respostas completas e parciais) foram avaliados. As respostas foram definidas pelos critérios da Organização Mundial da Saúde e submetidas a um comitê de revisão independente (IRC) cego. As diferenças nas avaliações entre o investigador e o IRC foram reconciliadas pelo patrocinador, cego para o braço de tratamento, de acordo com um algoritmo especificado. A sobrevivência foi avaliada com base em uma análise de não inferioridade.
Os dados demográficos de linha de base para pacientes com XELODA e 5-FU/LV são mostrados na Tabela 13.
Os endpoints de eficácia para os dois estudos de fase 3 são mostrados em e
Figura 3 Curva de Kaplan-Meier para sobrevivência geral de dados agrupados (estudos 1 e 2)
XELODA foi superior a 5-FU/LV para taxa de resposta objetiva no Estudo 1 e Estudo 2. A semelhança de XELODA 500mg e 5-FU/LV nesses estudos foi avaliada examinando a diferença potencial entre os dois tratamentos. A fim de assegurar que XELODA 500 mg tenha um efeito de sobrevivência clinicamente significativo, foram realizadas análises estatísticas para determinar a percentagem do efeito de sobrevivência de 5-FU/LV que foi retido por XELODA. A estimativa do efeito de sobrevivência de 5-FU/LV foi derivada de uma meta-análise de dez estudos randomizados da literatura publicada comparando 5-FU a regimes de 5-FU/LV que eram semelhantes aos braços de controle usados nesses estudos 1 e 2. O método para comparar os tratamentos foi examinar o pior caso (limite superior de confiança de 95%) para a diferença entre 5-FU/LV e XELODA 500mg, e mostrar que a perda de mais de 50% do 5- O efeito de sobrevivência FU/LV foi descartado. Foi demonstrado que a porcentagem do efeito de sobrevivência de 5-FU/LV mantido foi de pelo menos 61% para o Estudo 2 e 10% para o Estudo 1. O resultado combinado é consistente com uma retenção de pelo menos 50% do efeito de 5 -FU/LV. Deve-se notar que esses valores para efeito preservado são baseados no limite superior da diferença 5-FU/LV vs XELODA 500mg. Esses resultados não excluem a possibilidade de equivalência verdadeira de XELODA 500mg a 5-FU/LV (ver e Figura 3 ).
Câncer de mama
XELODA foi avaliado em ensaios clínicos em combinação com docetaxel (Taxotere®) e como monoterapia.
Em combinação com docetaxel
A dose de XELODA utilizada no ensaio clínico de fase 3 em combinação com docetaxel foi baseada nos resultados de um estudo de fase 1, em que uma gama de doses de docetaxel administradas em ciclos de 3 semanas em combinação com um regime intermitente de XELODA (14 dias de tratamento, seguido por um período de descanso de 7 dias). O regime de dose combinada foi selecionado com base no perfil de tolerabilidade dos 75 mg/m2 administrados em ciclos de 3 semanas de docetaxel em combinação com 1.250 mg/m2 duas vezes ao dia por 14 dias de XELODA 500 mg administrado em ciclos de 3 semanas. A dose aprovada de 100 mg/m2 de docetaxel administrada em ciclos de 3 semanas foi o braço de controle do estudo de fase 3.
XELODA em combinação com docetaxel foi avaliado em um estudo aberto, multicêntrico e randomizado em 75 centros na Europa, América do Norte, América do Sul, Ásia e Austrália. Um total de 511 pacientes com câncer de mama metastático resistente ou recorrente durante ou após uma terapia contendo antraciclina, ou recidivando durante ou recorrente dentro de 2 anos após a conclusão de uma terapia adjuvante contendo antraciclina foram inscritos. Duzentos e cinquenta e cinco (255) pacientes foram randomizados para receber XELODA 1250 mg/m2 duas vezes ao dia por 14 dias seguidos por 1 semana sem tratamento e docetaxel 75 mg/m2 como infusão intravenosa de 1 hora administrada em ciclos de 3 semanas. No braço de monoterapia, 256 pacientes receberam docetaxel 100 mg/m2 como uma infusão intravenosa de 1 hora administrada em ciclos de 3 semanas. Os dados demográficos dos pacientes são fornecidos na Tabela 16.
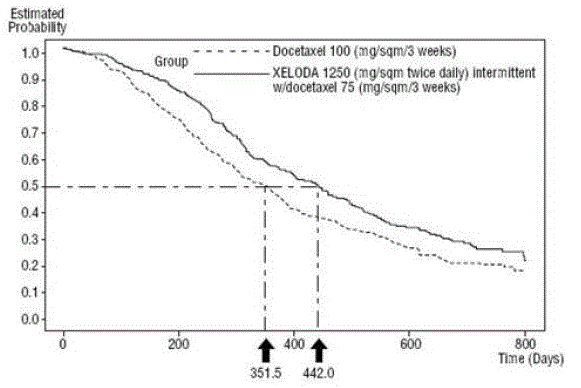
XELODA em combinação com docetaxel resultou em melhora estatisticamente significativa no tempo de progressão da doença, sobrevida global e taxa de resposta objetiva em comparação com a monoterapia com docetaxel, conforme mostrado em e Figura 5 .
Figura 4 Estimativas de Kaplan-Meier para Progressão do Tempo para a Doença XELODA e Docetaxel vs Docetaxel
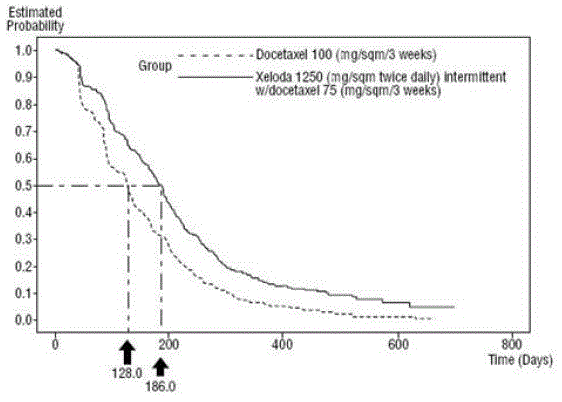
Figura 5 Estimativas Kaplan-Meier de Sobrevivência XELODA e Docetaxel vs Docetaxel
Monoterapia
atividade antitumoral de XELODA como monoterapia foi avaliada em um estudo aberto de braço único realizado em 24 centros nos EUA e Canadá. Um total de 162 pacientes com câncer de mama em estágio IV foram inscritos. O desfecho primário foi a taxa de resposta do tumor em pacientes com doença mensurável, com resposta definida como uma diminuição ≥50% na soma dos produtos dos diâmetros perpendiculares da doença bidimensionalmente mensurável por pelo menos 1 mês. XELODA foi administrado na dose de 1.255 mg/m2 duas vezes ao dia por 2 semanas, seguido por um período de descanso de 1 semana e administrado em ciclos de 3 semanas. As características demográficas e clínicas da linha de base para todos os pacientes (n=162) e aqueles com doença mensurável (n=135) são mostradas em . A resistência foi definida como doença progressiva durante o tratamento, com ou sem resposta inicial, ou recidiva dentro de 6 meses após a conclusão do tratamento com um regime de quimioterapia adjuvante contendo antraciclina.
As respostas antitumorais para pacientes com doença resistente ao paclitaxel e a uma antraciclina são mostradas em
Para o subgrupo de 43 pacientes que eram duplamente resistentes, o tempo médio de progressão foi de 102 dias e a sobrevida média foi de 255 dias. A taxa de resposta objetiva nesta população foi apoiada por uma taxa de resposta de 18,5% (1 CR, 24 PRs) na população geral de 135 pacientes com doença mensurável, que eram menos resistentes à quimioterapia (ver ). O tempo médio de progressão foi de 90 dias e a sobrevida média foi de 306 dias.
INFORMAÇÃO DO PACIENTE
XELODA® (zeh-LOE-duh) (capecitabina) comprimidos
Qual é a informação mais importante que devo saber sobre XELODA 500mg?
XELODA 500mg pode causar efeitos colaterais graves, incluindo:
- XELODA pode interagir com medicamentos anticoagulantes, como a varfarina (COUMADIN®). Tomar XELODA 500mg com estes medicamentos pode causar alterações na velocidade de coagulação do sangue e pode causar sangramento que pode levar à morte. Isso pode acontecer alguns dias após você começar a tomar XELODA 500 mg, ou mais tarde durante o tratamento, e possivelmente até 1 mês após você parar de tomar XELODA. Seu risco pode ser maior porque você tem câncer e se tiver mais de 60 anos de idade.
- Antes de tomar XELODA, informe o seu médico se estiver a tomar varfarina (COUMADIN) ou outro medicamento anticoagulante.
- Se você tomar varfarina (COUMADIN) ou outro anticoagulante semelhante à varfarina (COUMADIN) durante o tratamento com XELODA, seu médico deve fazer exames de sangue com frequência para verificar a rapidez com que seus coágulos sanguíneos durante e após a interrupção do tratamento com XELODA. Seu médico pode alterar sua dose do medicamento para afinar o sangue, se necessário.
Ver “Quais são os possíveis efeitos colaterais do XELODA 500mg?” para obter mais informações sobre os efeitos colaterais.
O que é XELODA?
XELODA é um medicamento de prescrição usado para tratar pessoas com:
- câncer do cólon que se espalhou para os linfonodos na área próxima ao cólon (estágio C de Dukes), após a cirurgia.
- câncer de cólon ou reto (colorretal) que se espalhou para outras partes do corpo (metastático), como seu primeiro tratamento de seu câncer neste estágio.
- câncer de mama que se espalhou para outras partes do corpo (metastático) juntamente com outro medicamento chamado docetaxel após o tratamento com alguns outros medicamentos anticancerígenos não funcionaram.
- câncer de mama que se espalhou para outras partes do corpo e não melhorou após o tratamento com paclitaxel e alguns outros medicamentos anticancerígenos, ou que não pode receber mais tratamento com certos medicamentos anticancerígenos.
Não se sabe se XELODA 500mg é seguro e eficaz em crianças.
Não tome XELODA se você:
- tem problemas renais graves
- são alérgicos à capecitabina, 5-fluorouracil ou a qualquer um de seus ingredientes em XELODA. Consulte o final deste folheto para obter uma lista completa dos ingredientes de XELODA.
Fale com o seu médico antes de tomar XELODA se não tiver certeza se tem alguma das condições listadas acima.
Antes de tomar XELODA, informe o seu médico sobre todas as suas condições médicas, incluindo se você:
Ver “Qual é a informação mais importante que devo saber sobre XELODA?”
- teve problemas cardíacos
- tem problemas renais ou hepáticos
- lhe foi dito que lhe falta a enzima DPD (dihidropirimidina desidrogenase).
- estão grávidas ou planejam engravidar. XELODA 500mg pode prejudicar o feto. Seu médico deve fazer um teste de gravidez antes de iniciar o tratamento com XELODA. Informe imediatamente o seu médico se engravidar ou se pensa que pode estar grávida durante o tratamento com XELODA.
- Mulheres que podem engravidar devem usar métodos contraceptivos eficazes durante o tratamento e por 6 meses após a dose final. Converse com seu médico sobre as opções de controle de natalidade que podem ser adequadas para você durante o tratamento com XELODA.
- Machos que têm parceiras que podem engravidar devem usar métodos contraceptivos eficazes durante o tratamento e por 3 meses após a dose final.
- está amamentando ou planeja amamentar. Não se sabe se XELODA passa para o leite materno. Não amamente durante o tratamento com XELODA 500mg e por 2 semanas após a dose final.
Informe o seu médico sobre todos os medicamentos que você toma, incluindo medicamentos prescritos e de venda livre, vitaminas e suplementos de ervas. XELODA pode afetar a forma como outros medicamentos atuam, e outros medicamentos podem afetar a forma como XELODA atua.
Conheça os medicamentos que você toma. Mantenha uma lista deles para mostrar ao seu médico e farmacêutico quando receber um novo medicamento.
Como devo tomar XELODA 500mg?
- Tome XELODA exatamente como seu médico lhe disse para tomá-lo.
- Seu médico lhe dirá quanto XELODA deve tomar e quando tomá-lo.
- Tome XELODA 2 vezes ao dia, 1 vez de manhã e 1 vez à noite.
- Tome XELODA 500mg dentro de 30 minutos após terminar uma refeição.
- Engula os comprimidos de XELODA 500mg inteiros com água. Não faça esmagar ou cortar os comprimidos de XELODA 500mg. Se não conseguir engolir os comprimidos de XELODA inteiros, informe o seu médico.
- Seu médico pode alterar sua dose, interromper temporariamente ou interromper permanentemente o tratamento com XELODA 500mg se você desenvolver efeitos colaterais.
- Se você tomar muito XELODA, ligue para o seu médico ou dirija-se ao pronto-socorro do hospital mais próximo imediatamente.
Quais são os possíveis efeitos colaterais do XELODA?
XELODA pode causar efeitos colaterais graves, incluindo:
Náuseas e vômitos são comuns com XELODA. Se você perder o apetite, sentir-se fraco e tiver náuseas, vômitos ou diarreia, poderá ficar rapidamente desidratado.
Pare de tomar XELODA e ligue para o seu médico imediatamente se você:
Se a contagem de glóbulos brancos for muito baixa, você tem um risco aumentado de infecção. Ligue para o seu médico imediatamente se desenvolver febre de 100,5 oF ou mais ou tiver outros sinais e sintomas de infecção.
- Ver “Qual é a informação mais importante que devo saber sobre XELODA?”
- Diarréia. A diarreia é comum com XELODA e às vezes pode ser grave. Pare de tomar XELODA e ligue para o seu médico imediatamente se o número de evacuações que você tem em um dia aumenta em 4 ou mais evacuações do que o normal para você ou evacuações à noite. Pergunte ao seu médico sobre quais medicamentos você pode tomar para tratar sua diarreia. Se você tiver diarreia sanguinolenta grave com dor abdominal intensa e febre, pare de tomar Xeloda 500mg e ligue para o seu médico ou dirija-se imediatamente ao pronto-socorro do hospital mais próximo.
- Problemas cardíacos. XELODA pode causar problemas cardíacos, incluindo: ataque cardíaco e diminuição do fluxo sanguíneo para o coração, dor no peito, batimentos cardíacos irregulares, alterações na atividade elétrica do seu coração observadas em um eletrocardiograma (ECG), problemas com o músculo cardíaco, insuficiência cardíaca e morte. Pare de tomar XELODA 500mg e ligue para o seu médico ou dirija-se ao pronto-socorro do hospital mais próximo imediatamente se tiver quaisquer novos sintomas de um problema cardíaco, incluindo:
- dor no peito
- tontura
- falta de ar
- tontura
- Perda de muito líquido corporal (desidratação) e insuficiência renal. A desidratação pode ocorrer com XELODA 500mg e pode causar insuficiência renal súbita que pode levar à morte. Você está em maior risco se tiver problemas renais antes de tomar XELODA 500 mg e também tomar outros medicamentos que podem causar problemas renais.
- vomitar 2 ou mais vezes por dia.
- só conseguem comer ou beber um pouco de vez em quando, ou não conseguem comer nada devido à náusea.
- tem diarreia. Veja “diarréia” acima.
- Reações graves na pele e na boca.
- XELODA 500mg pode causar reações cutâneas graves que podem levar à morte. Informe o seu médico imediatamente se desenvolver uma erupção cutânea, bolhas e descamação da pele. O seu médico pode dizer-lhe para parar de tomar XELODA se tiver uma reação cutânea grave. Não tome XELODA 500mg novamente se isso acontecer.
- XELODA 500mg também pode causar síndrome de “mão e pé”. A síndrome das mãos e pés é comum com XELODA 500mg e pode causar dormência e alterações na sensibilidade das mãos e pés, ou causar vermelhidão, dor, inchaço das mãos e pés. Pare de tomar XELODA e ligue para o seu médico imediatamente se tiver algum desses sintomas e não puder realizar suas atividades habituais.
- A síndrome da mão e do pé pode levar à perda de impressões digitais que podem afetar sua identificação.
- você pode ter feridas na boca ou na língua ao tomar XELODA. Pare de tomar XELODA 500mg e ligue para o seu médico se tiver vermelhidão dolorosa, inchaço ou úlceras na boca ou língua, ou se tiver problemas para comer.
- Aumento do nível de bilirrubina no sangue e problemas no fígado. O aumento da bilirrubina no sangue é comum com XELODA 500mg e às vezes também pode ser grave. O seu médico irá avaliá-lo quanto a estes problemas durante o tratamento com XELODA.
- Diminuição da contagem de glóbulos brancos, plaquetas e glóbulos vermelhos. Seu médico fará exames de sangue durante o tratamento com XELODA para verificar suas contagens de células sanguíneas.
Pessoas com 80 anos de idade ou mais podem ter maior probabilidade de desenvolver efeitos colaterais graves ou graves com XELODA.
Os efeitos colaterais mais comuns de XELODA 500mg incluem:
- diarréia
- dor na área do estômago (abdominal)
- síndrome mão e pé
- fraqueza e cansaço
- náusea
- vomitou
- quantidades aumentadas de produtos de degradação dos glóbulos vermelhos (bilirrubina no sangue
Reações alérgicas graves podem ocorrer com XELODA. Informe o seu médico se você já teve uma reação alérgica à capecitabina ou 5-fluorouracil. Ver “Não tome XELODA se você:”. Pare de tomar XELODA 500mg e ligue para o seu médico imediatamente ou vá a uma sala de emergência se tiver algum dos seguintes sintomas de uma reação alérgica grave:
- vergões vermelhos com coceira na pele (urticária)
- vermelhidão da pele
- inchaço do rosto, lábios, língua ou garganta
- irritação na pele
- coceira
- dificuldade para engolir ou respirar
XELODA 500mg pode causar problemas de fertilidade em mulheres e homens. Isso pode afetar a capacidade de ter um filho. Converse com seu médico se tiver dúvidas sobre fertilidade.
Estes não são todos os possíveis efeitos colaterais do XELODA.
Ligue para o seu médico para aconselhamento médico sobre os efeitos colaterais. Você pode relatar efeitos colaterais ao FDA em 1-800-FDA-1088.
Como devo armazenar o XELODA?
- Armazene XELODA 500mg em temperatura ambiente entre 68°F a 77°F (20°C a 25°C).
- Mantenha XELODA 500mg em um recipiente bem fechado.
- Pergunte ao seu médico ou farmacêutico como jogar fora com segurança qualquer XELODA não utilizado.
Mantenha XELODA e todos os medicamentos fora do alcance das crianças.
Informações gerais sobre o uso seguro e eficaz de XELODA.
Os medicamentos às vezes são prescritos para outros fins que não os listados em um folheto de informações ao paciente. Não use XELODA para uma condição para a qual não foi prescrito. Não dê XELODA 500mg a outras pessoas, mesmo que tenham os mesmos sintomas que você. Pode prejudicá-los. Você pode pedir ao seu farmacêutico ou profissional de saúde informações sobre o XELODA 500mg que foi escrito para profissionais de saúde.
Quais são os ingredientes de XELODA 500mg?
Ingrediente ativo: capecitabina
Ingredientes inativos: lactose anidra, croscarmelose sódica, hidroxipropilmetilcelulose, celulose microcristalina, estearato de magnésio e água purificada. O revestimento de película de pêssego ou pêssego claro contém hidroxipropilmetilcelulose, talco, dióxido de titânio e óxidos de ferro sintéticos amarelos e vermelhos.
Para obter mais informações, acesse http://www.gene.com/patients/medicines/xeloda 500mg ou ligue para 1-877-436-3683.
Esta informação do paciente foi aprovada pela Food and Drug Administration dos EUA.